There are several details here that can drastically alter the calculations. First, we can either assume that the ethanol vapor entering the condenser is at its boiling point or needs to be cooled to its boiling point before it can condense. Next, will we be running at atmospheric pressure or at vacuum.
I have taken the liberty of assuming that the condenser duty is calculated for condensing ethanol at its boiling point and at atmospheric pressure. This simplifies things.
Now, calculate the heat required to condense the gaseous ethanol:
Qdot = ndot*Hvap
Qdot = Heat Duty Required
ndot = Molar Flow Rate of Gaseous Ethanol
Hvap = Specific Molar Heat of Vaporization for Ethanol
Molar flow rate is simply converting lpm to mole/min using density and molecular weight (i will provide my written calculations)
This gives ~ 250,000 kJ/min of Required Heat Duty JUST FOR CONDENSATION, NO COOLING.
Now, things begin to get extremely convoluted, complicated and open to design optimization.
The first thing we want to do is see what kind of temperature change we would see for 10 lpm of water encountering this heat duty.
Qdot = mdotCp(Th-Tc)
Qdot = Same as above.
mdot = mass flow rate of water per minute
Cool = Heat capacity of water in (kJ/g*C)
Th, Tc = Temperature (in Celsius) of water exiting condenser and water entering condenser respectively.
For a 10 lpm Stream of water, the change in temperature is over 5000C! So clearly, we need to explore a different avenue.
Fortunately, there is an alternative way forward:
Qdot = UALMTD
U = Overall Heat Transfer Coefficient
A = Area available for Heat Transfer
LMTD = Log Mean Temperature Difference
Hoo boy, it just got even more complicated…
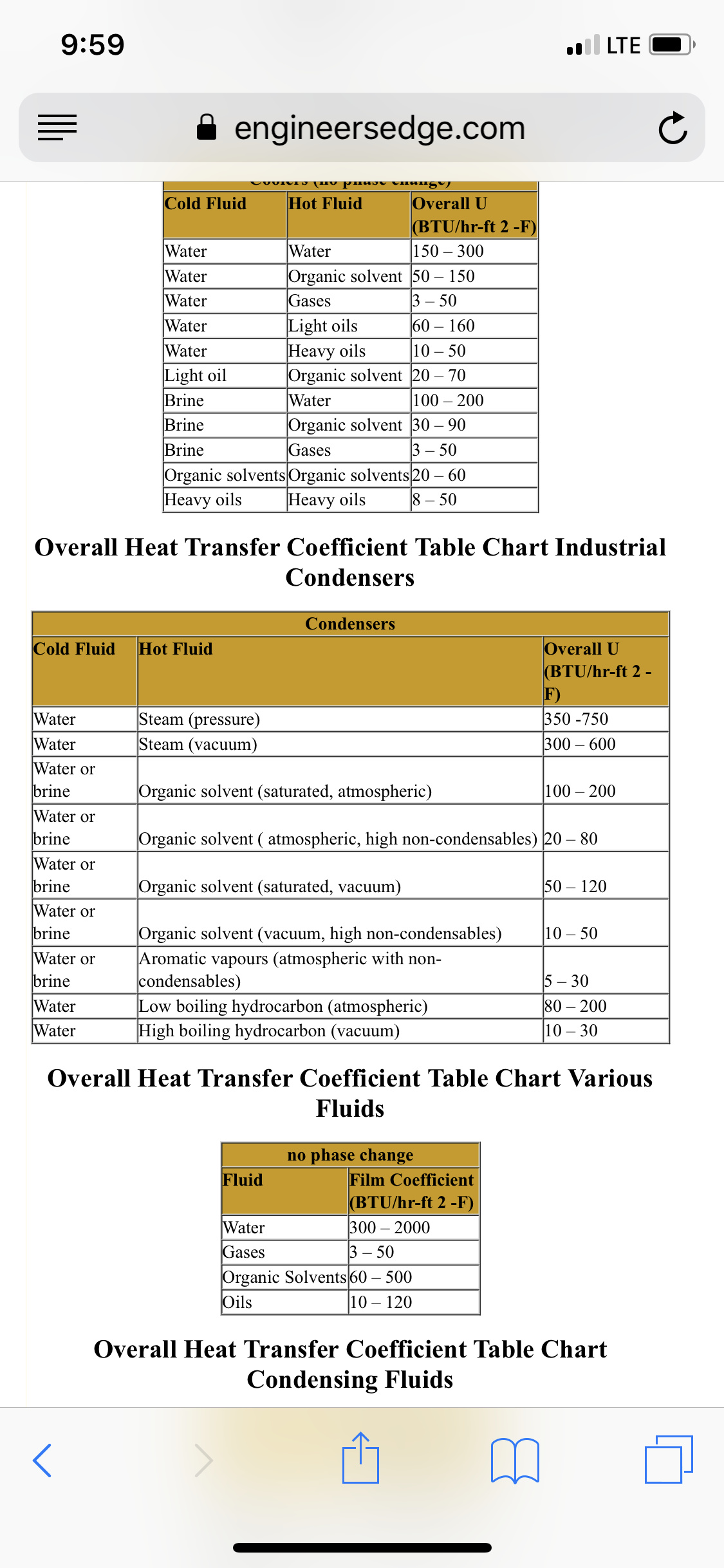
U is a difficult to calculate number that has ruined the life of many chemical engineers, but there are relatively decent approximations for given situations, in this case to condense an “organic solvent” with a cooling water stream can be found in the table below. So we will assume the extreme values and end up with a range of surface areas.
LMTD is essentially the difference between the difference of the cold stream and gaseous ethanol and the hot stream and gaseous ethanol, google it if you’re interested. BUT THIS MEANS I will have to assume the exit temperature of the cooling water—let’s say it’s 45 Celsius.
So now A = Qdot/(U*LMTD)
For the worst-case scenario A1 = 284.15 meters squared
A2 = 71.04 meters squared
Both insanely large areas, but 378 L/min is equivalent to nearly 430,000 kg/day of ethanol!!
Using all this data we can back calculate to see the required flow rate of water to cool this flow rate of ethanol using the second provided equation.
Anyways, anything confusing that I can clarify on, here?