Theoretically, and practically, I have come to learn that the heat transfer in falling film is sensitive to the characteristic length, i.e. inner diameter of the tube/column. Holding viscosity constant, the diameter and the mass flow rate are driving parameters… Designers of falling film evaporators are keen on getting the right Gamma value which is a ratio of 4 times mass flow to unit length (characteristic length). Single pass evaporators are tough to get good Gamma values… That’s why you see recycle a lot.
Great calcs above, reminds me of my unit ops courses… Fellow Chem Eng here…
2 Likes
Is the “goal” to “collide” solvents under constant pressure and temps and the resulting “free energy” causes precipitation?
I typed out this explanation only to realize it doesn’t really answer your question, I’m gonna post it for posterity and then get to your question: The free energy of a system in which crystallization is occurring can be understood in the following way: there is both a surface energy of a nucleating particle and volume free-energy.
The Gibbs Free Energy, then, is a combination of these two factors and then becomes a function of Radius. G(r) = volume-coefficientsr^3 + surface-coefficientsr^2
At a certain critical radius, r*, the Gibbs free energy begins to decrease as radius of the nucleate body increases, as Gibbs free energy decreases the crystallization becomes more thermodynamically favorable and when it decreases below 0 (becomes negative) the crystals begin to grow spontaneously.
Edit: Picture for clarity.
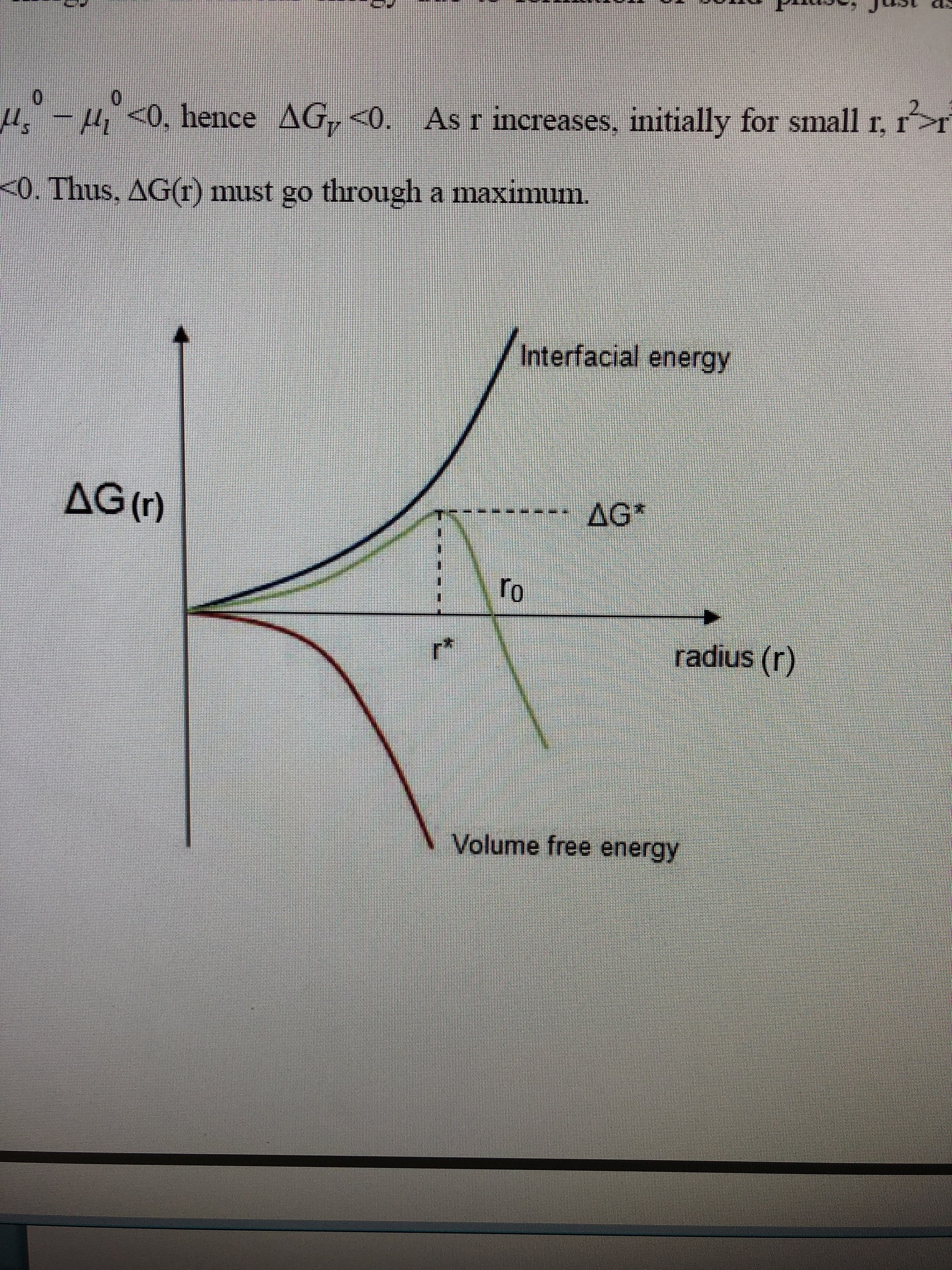
2 Likes
The “goal” is indeed to collide molecules under controlled pressure and temps, because if you can increase the size of the nucleate by controlling these factors then the critical radius (r*) is surpassed and you create spontaneous crystal growth.
In equation 29, below you’ll see the term (Tm-T) has an influence on rate of solidification…this means that as the difference between the melting temperature ™ and the temperature of the system increase—I.e. if you cool the system far below its melting point—the rate of crystallization will increase but the term in the middle will decrease. So there is essentially a sweet spot where the temperature is cold but not too cold.
This can be seen in the graph below.
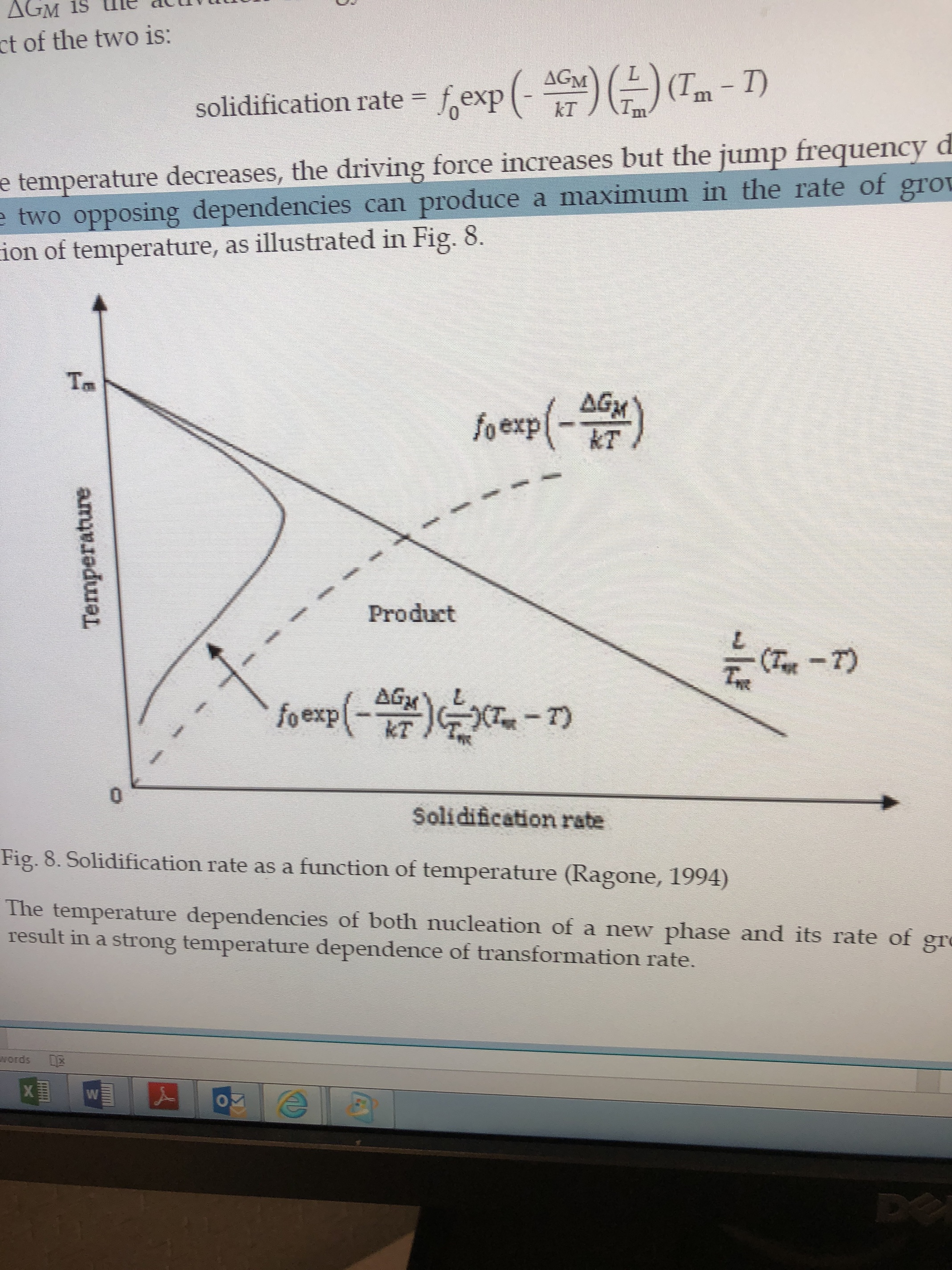

Now this continues to complicate itself, and I know my explanation is not the greatest thing ever, but I will continue.
Now starting to diverge from free energy a little. Crystallization on a macro scale is entirely controlled by super saturation. Super saturation can be induced by increasing the temperature of your solvent so that more solute falls into solution then cooling so it falls out.
As the degree of super saturation increases the rate of nucleation increases, the growth rate increases and the crystal size decreases. All things we’d want.
SO, real world example. Heat your high cannabinoid distillate up as much as you can without ruining your product, hold it as long as safely possible, then cool. Cool to different temperatures to determine the ‘sweet spot.’

2 Likes
It’s all about supersaturating your solvent and then cooling to an ideal temperature that has to be determined experimentally.
3 Likes
Awesome thank you!
@TheGratefulPhil
I have a question about boiling points. If you were observing a compound in a boiling flask with the pressure held constant at ¾ of one micron, what criteria would you use to determine if the compound is boiling or not? I observe condensate on the glass surface of the unfilled cold finger as low as 120C as measured between the glass and heating mantle at these vacuum levels. However it takes a very long time to accumulate much at this temp. I have used 120C as a data point for a boiling temp/pressure curve. However I have no idea how others judge when a boil occurs at such pressure.
What indicator would you use to define a “boil” in such a circumstance which then is mapped to a temp and pressure?
I also have unanswered questions about women. It has been decades I am not sure I get it yet? That is a different post though…![]()
I’m not sure if you’ve heard of the McCabe-Thiele graphical method, but it’s something used to graphically determine the number of trays and feed location of a distillation column.
Anyways, Thiele, being the genius he was, devised a way to determine boiling points with something now called a ‘Thiele Tube.’ I quick google found this organic chemistry lab experiment from Calgary
Essentially you will have to utilize this method at varying pressures. I would begin with atmospheric and very gradually, VERY GRADUALLY pull vacuum. You will almost certainly need someone to custom make this piece if you expect it to hold vacuum and perhaps you can just pull vac in the micro-tube.
Anyways, this is the method I’m most familiar with in terms of determining boiling point, the determination at reduced pressures will require a customized version ![]()
2 Likes
As for women, I don’t even know how I have a girlfriend man. Engineers ain’t the right ones to ask about women ![]()
4 Likes
That tube is prettty smart but it relies on atmospheric pressure as a reference. The definition has to do with the vapor point equaling pressure. So how do
I proceed when there is no system pressure as a practical matter? I can’t envision how the idea could be adapted to an environment where there is effectively no atmosphere?
Thanks for the page. It is informative.
I was thinking something along the lines of using the same setup but evacuating the micro tube used to hold the sample. So, if the micro tube and the inverted capillary tube were subject to reduced pressure, their boiling point / vapor pressure could be determined. No?
A glass rod 8mm in diameter fits a thermometer adapter. A three neck flask could be used. The glass rod stuck into the bottom of a large flask immersed in puddle. Thermometer opposing neck into the bottom also but not touching. Immersed in puddle. Vacuum adapter center neck.
Without vacuum and with the fluid compound hot enough to flow it could be drawn up into the glass tube from the puddle past a small one way vacuum valve and sealed shut. Then the puddle would be pulled down to vacuum.
Apply heat then until the glass rod is evacuated totally. Stop heat and monitor tube and temp of puddle. The moment the compound enters back into the tube the temp reading is agreed then to be the boiling temp at the indicated pressure.
Does this sound like it mirrors the intent of the link you posted? The Thiel Tube?
By the way, is every single item in a lab named after the guy who cobbled it together in the first place? ![]()
1 Like
Do you have any shorthand formulas for determining miltipath effect and longitudinal diffusion? Trying to determine optimal load rates for multiple absorbents. TY
2 Likes
not in my lab…but they all get names…otherwise sending a minion off to assemble apparatus becomes waaay too complicated.
First step of my SOP’s used to be “assemble the required equipment”. As everything was cobbled together/assembled from tri-clamp parts, that first step could be a bitch.
At this point most of the dohicky’s have names, and stable parts lists. Getting their pictures & component parts into the SOP’s is still a work in progress.
1 Like
I will probably need a little more context than that. You’re trying to load something onto an adsorbent? From gas or liquid phase? Are you simultaneously trying to strip the adsorbed molecules into another phase? The same phase?
What kind of packing? Vertical or horizontal column? What kind of flow rates and pressures are you generally operating at? Temperatures?
Are there any solvents/gasses in the mixture that you know of for sure?
I dont want you to give away your process but a little bit of context might help
Fellow engineer (mechanical) here, process optimization really turns my crank, though most of my experience is with non-physical systems. I think a thread like this is a great idea, @TheGratefulPhil!
My current pondering is about how to practically go about optimizing boiling point depression through applying varying levels of vacuum in a falling-film evaporator to:
- ensure the highest recovery throughput possible, and
- minimize energy requirements, (in that order).
Working numbers:
- Heat source is Saturated steam at 250 deg F / 120 deg C (approx 14.7psi / 1bar)
- Cooling source is some form of closed-loop glycol mix at approximately 42 F / 5.6 deg C
- Input solvent/oleoresin mix is going to be somewhere in the range of -40 deg C, but will be pre-heated to something approaching BP by pre-heating coils before it hits the main evaporation system.
Let’s assume I’ve got effectively unlimited (but not zero-cost) heat and cooling sources, and a vacuum source able to hold any realistic vacuum I want, so as minimize the variables that can bork this (assume spherical cows, etc). This allows me to set the boiling point of the azeotrope wherever I want.
Rough estimates of boiling points of an ethanol-water azeotrope (ignoring effect of the resin on the BP) come to:
- 6.6 deg C @ 26 torr
- 50 deg C @ 250 torr
[ using formula (1/((1/351.25)-((8.314/38560)*LN( TORR /760))))-273.15 as derived from calculator at Boiling Points of Ethanol and Water ]
Per 100L/hour of recovery, every 10 deg C of difference between the input and the boiling point adds at least 560W of heating, and some to be determined but likely lesser amount of cooling.
The realistic extremes I could work with with this setup are:
- 760 torr (atmospheric), with a BP of 78 deg C, and a delta of 72 deg C between the vapour and the condenser
- 25 torr, with a BP of 6.6 deg C, and a delta of 1 deg C between the vapour and the condenser
My (limited and probably over-generalized) understanding of thermodynamics suggests that:
- the fastest/most efficient vapourization of the input stream will happen with the highest delta between the heat source and the input stream, implying the deeper the vacuum the better.
- the fastest/most efficient condensation of the solvent stream will happen with the highest delta between the cooling source and the vapour stream, implying that atmospheric pressure with the highest BP is going to be best.
Optimizing either part of the system is reasonably trivial. Optimizing them as a combined unit is substantially more difficult.
The above makes me think that I should set the vac level so as to drop the BP to somewhere in the middle, say around 42 deg C @ 170 torr. Or even 63 deg C @ 420 torr (heh) to put the BP right in the middle between the heat source and cooling source temps.
I’d appreciate any theoretical or practical knowledge, research papers, or other input that applies here.
And yes, I do plan on playing with this empirically, but until the equipment is manufactured that’s hard to do, and investors want to see operations and revenue flowing as fast as possible once it’s all delivered.
3 Likes
How do you propose to vary the vacuum depth?
1 Like
With a vacuum controller/regulator.
To clarify, I don’t mean to vary it during the process, but rather playing with it up front to determine the optimal vac level, which will become the setpoint in future.
2 Likes
I believe you will find attempting to control the vacuum levels with a regulator is impractical. The vacuum is dictated by many factors not the least of which is the composition of the compound itself. Attempting control of this is more of an engineering challenge than it might appear.
The only way to control vacuum is to bleed off vacuum down to a set point. A regulator therefor is an intentional “leak” in system vacuum so as to maintain a set point. It is my experience that the vacuum pumps we use cannot achieve as deep of a vacuum as is optimum in most cases so trying to regulate vacuum levels via a leak down regulator or trying to regulate the vacuum with a variable frequency drive on the motor is a moot point. The optimum vacuum will be revealled to be the maximum blank off vacuum your system can generate.
Vacuum levels indicate many things in a system besides internal pressure. One thing you can monitor is the volitiles still present in the boil and this is useful BUT the vast majority of designs do not leverage this information currently. You speak of falling film which has its own niche in refinement and serves a specific purpose.
Overall though the idea of reducing boiling points can be achieved in a few ways and are well known the idea of system pressures and why we see what we see seems a bit less well known. The vacuum level is not set as simply as a heat control even though pressure and heat inside a closed system are one and the same from a certain point of view. Vacuum level just measures density and not the energy directly. When you have say a compound with 50% volitiles all boiling off the vacuum levels will be low compared to when those have nearly depleted in the system. So vacuum level can tell you the engineer just when the puddle is getting down to the good stuff. I use this as an indicator to inform me when the volitiles are gone and then can adjust my temperature and prep the cold finger in my unit for cannabinoid harvest.
There is no case in which the ideal vacuum is less than my pump can generate and my instrumentation is accurate down to one micron of system pressure. When the vacuum is above about 20 microns I will generate volitiles like terpenes. Once the vacuum pulls down below that I can be certain of the next fraction and also then be certain of the composition and adjust my temp accordingly. I typically see one micron system pressures once the sweet spot hits and if the pump is optimum like when fresh oil is put in. This is the other problem with vacuum - pump contamination of oil but that is a different post.
I do not have a collegiate background but I did graduate as a Madras White Buffalo albeit with a D- average lolz. You have to keep language simple for me. ![]() The educated types on the forum tolerate me because they needed a whacky side kick and I kept showing up… I think you have an enormous amount of knowledge to draw from as do all folks and your pool of knowledge includes college level work. Normally by this point in a reply I work in a jab or two at the Phd types and take pot shots at education in general but I woke up in a good mood. Plus @Shadownaught seems like a nice Phd type so maybe there is hope for you guys?
The educated types on the forum tolerate me because they needed a whacky side kick and I kept showing up… I think you have an enormous amount of knowledge to draw from as do all folks and your pool of knowledge includes college level work. Normally by this point in a reply I work in a jab or two at the Phd types and take pot shots at education in general but I woke up in a good mood. Plus @Shadownaught seems like a nice Phd type so maybe there is hope for you guys? ![]() (could not help it!)
(could not help it!)
Good luck and welcome to this place on the net. I believe this blog to host the finest minds on the subject there are on the topics. I admire the ambition of your introductory post and if I can help at all I will.
1 Like
Oh I’m definitely a level 9000 super genius…/s
1 Like