Would a pressure canner with steam at 15psi or so be of any benefit over decarb at Atmos?
No, and that would likely put too much water emulsion in the stuff.
3 Likes
Do you know the mechanism for MgO catalyzing this reaction?
1 Like
If you want to keep your terms why aren’t you just vacuum distilling the terpenes off at decarboxylation temps?
1 Like
For the same reason I extract cold rather than winterize. Fewer steps.
Or at least that’s the rational for trying…
4 Likes
How does that reduce steps when you will have to remove terps in a later step anyway?
Decarbed crude in a pen (hopefully with retained terps)
Devol into lungs.
Fewer steps…
1 Like
What is your crude testing at?
In house potency is high 70’s or low 80’s.
Pretty sure the BHO had 3rd party in the 77-78% thc range.
That COA is probably 3years old, and the EHO is a little over a year old at this point… those numbers are from memory, so calling them more than guesses is probably a stretch.
Why?
Just a little repelled at the thought of non-distillate in a cartridge. But then again, I’ve never seen you’re stuff
Personal meds. From R&D work.
Tasted like hell in pods I was told to fill.
Tasted & worked great in everything else I tried it in. Although I never could get a 3g charge to not leak at about 1/2 done.
Turns out same was true for the outsourced hydrocarbon “sauce” pods.
Who releases a pod device without wattage control (temp control would be better)?
The idea of turning 70% thc to 3% thc by “distilling” it, and not really knowing what it is that became of the thc leaves me avoiding distillate I didn’t make AND generate a chromatogram on.
One mans trash, another mans vape juice I guess.
Couldn’t sell it, cause R&D is explicitly straying from SOP. in OR your SOP is supposed to be static & validated.
3 Likes
You mention that pressure does not affect decarb rxn kinetics. I want to point out that if you apply enough pressure to raise the boiling point of terps, THC, CBD, etc… you can increase the decarb temperature in turn speeding up the decarb without risking the loss of these compounds. I found this system online, its a bigger version of something I used at my university’s chem lab:
https://syrris.com/product/titan/
It doesn’t apply pressure by blanketing an inert gas, it uses large syringe pumps and a valve to create backpressure. But it would apply pressure and heat in order to decarb the extract and retain the terps if my logic is correct based on the science of the previously linked lacy system.
Increasing the decarboxylation temperature speeds up decarboxylation? And how could that work without any headspace? CO2 is an inert gas and has to come out somewhere. Do you mean increasing pressure on a liquid increases its temperature? That isn’t really true, although maybe there is more energy applied, which could speed up decarboxylation. In any case the temperature would not be affected and you would create a very high pressure scenario by impinging the liquid in a syringe type positive displacement system. You may be correct about losing fewer of the volatile compounds, but increasing the energy input would also speed up any thermal decomposition reactions.
2 Likes
Increasing pressure raises the boiling point, so you would not lose volatile compounds as easily compared to running at atmospheric pressure. Relative to this topic if you ran at the same temp, but under pressure, you should retain terpenes because they would not be lost as easily. The CO2 off gas would be held as a slug/bubble in the flow path and exit out the end of the system, it is a flow synthesis reactor so the reactants are pushed through the reactor at high pressure. At the end of the reactor after whatever valve is controlling back pressure, the CO2 would just vent out with the decarbed product.
Raising pressure raises boiling points, yes, but it doesn’t speed up decarboxylation.
1 Like
my initial comment was that if you raised pressure to increase boiling points and increased temp, it would increase rxn rate 2 fold for every 10C, Arrhenius equation
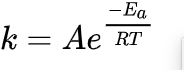
1 Like
I take your meaning, but decarboxylation is a pseudo first order reaction, so pressure doesn’t really affect it. You can decarb under pressure or vacuum if you want, but the temperature and rate remain the same, regardless… unless you add a catalyst like MgO.
5 Likes
I think you are missing the point I am making about speeding up the decarb in relation to terps-
Point 1- increasing the temp speeds up the decarb.
Point 2- if you also increase the pressure, thereby increasing boiling points, you would not lose as many volatile compounds such as terps? (questionable)
I think if you break down these points, I will better understand more any arguments against my logic that you can offer!
I’m just trying to see if these flow chemistry reactors can offer any benefit in the decarb process to better preserve secondary compounds such as terpenes. I was told these flow chemistry systems are highly customizable to run at different degrees of temp/pressure. If you run it with a pressure level greater than what is given off by the decarb rxn- any off gassing from a reaction is pushed out the end of the system, so CO2 would just flow out the back end.
Edit: Is terpene loss during decarb related to oxidation or thermal breakdown (or both?!) These flow systems run under inert atmosphere, so oxidation relating to O2 is limited.
Edit #2: Because there is only one way out of the reactor, anything that boils off during the decarb, would exit into the same container that the activated extract would collect into, putting it back in suspension. If this vessel had an open arm and a size excluding filter on it, you could keep terpenes in solution and let the CO2 escape! I was told you can also put a cooling cycle immediately following the heated reactor to condense anything volatile that escaped solution (terpenes). As long as you don’t go too cold, CO2 won’t recondense into solution.
CO2 is about 9-14 A^2 in size terpenes are about 30-75 A^2 in size. Just put a filter that lets CO2 escape and trap the terps in teh collection vessel.
Thanks Photon_noir! or anyone else that can offer insight into this application I am working on for preserving terps during decarb.
7 Likes
Yes, it makes more sense when you separate and organize the causes and effects! Thermal decomposition of terpenes can happen at high temperatures and pressures, so there is a balance to strike… ultimately, you might be better off using the system in reflux (of terpenes) under an unpressurized inertgas blanket, rather than trying to force the terpenes to remain in liquid state the entire time by pressurizing.
You probably cannot achieve cold sufficient to condense CO2 without pressure, correct.
It is also fairly simple, for that reason, to reflux condense the terpenes while CO2 escapes.
6 Likes
Would it not make more sense to decarb under vacuum if going straight from crude to disty? Stil as many volotiles as possible? I’m thinking roto under vacuum decarb might be better than an Atmos decarb?
1 Like