Hi,
i recently aquired a passive closed loop system for bho extraction, works like a charm, even better i also got a 2nd System for free.
But since dry ice is quite hard to get here and i can only work with ice + salts, i would like to convert it into an (semi) active closed loop system, to speed up the recovery (and as a bonus maybe be able a loop).
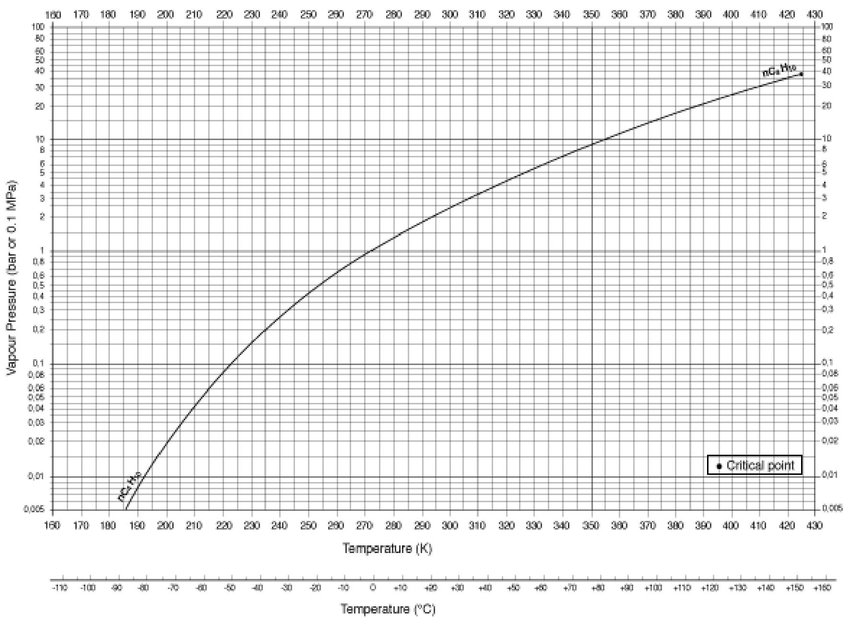
According to this graph:
https://encyclopedia.airliquide.com/butane#properties
Butane @3 bars should allready condens at ~302k, @2 bars it should condens at about 291 k, which is also quite near to the Clausius-Clayperon equiation result and this calculator:
https://www.omnicalculator.com/chemistry/boiling-point
I’m not looking to modify the system to a 300 psi system, but rather a hybrid system, keeping the collection vessel in a cold bath (salt/ice), and apply a pressure ~3 bars to the recovery vessel to speed up the recovery time to about <30 min.
I was thinking about using a small piston pump for this, any idea which one to use, or better use a diaphragm one, or what specs to look out for?
Alternativly i could imagine, to just connect an air compressor to the system, and pressurize the whole system.
Also i would like to add a CRC collum to the system, can i just pack silica/activated charcoal under the bud or do i really need to add an extra colum? Any experience on how it does on dewaxing, or do i really need to dewax with dry ice in the jacketed colum/winterisation?