Would a turbovap work?
It should. But it look like this is fancier and rather made for larger volumes of several dozen of ml.
The small centrifuge are used in general for small volumes <1.5 ml, which is far sufficient for the GC analysis. Plus de centrifugal force drags the sample to bottom of the tube, which makes the subsequent reaction with the limited <0.1 ml of derivatization agent much more convenient…
2 Likes
I also recommend baking the hardware out in a vacuum oven for an hour or two afterwards to clear out any solvent but that’s pretty much all there is to it.
1 Like
Understood. Hopefully I don’t fuck it up ![]()
One of the reasons why I leave my 10” fire proof fan, hooked up to variac, on at all times; just in case…
![]()
![]()
![]() well it looked something like that when i got it…
well it looked something like that when i got it…
Assuming you mean to not cut and refit the connections?
Speaking of inlet/outlet; what liners are you all using for your split/split less inlets? @SidViscous @Dr_Jebril @Chaboes @bigbone @thesk8nmidget
What… no way…

Well, this certainly won’t pass inspection…

Left to right: hydrogen generator, nitrogen generator, zero air generator (spent the last 3 hours taking it apart, replacing fittings, gaskets, down to 1 leak in a copper line which I’ll probably replace with a push to connect… ![]() ). Not pictured: 1 gallon ultra quiet harbor freight air compressor, so far, I’m a fan.
). Not pictured: 1 gallon ultra quiet harbor freight air compressor, so far, I’m a fan.

Pardon the mess. RO valve to a splitter which goes off to glove box and GC.

Replaced with RO valve, worked better than the one I had before. N2 line with valve going to glovebox

Top is h2 and bottom is N2
No leaks that I could detect at any taped fitting, nor the push connect; though, the original ones i used, which were black and “pneumatic” weren’t air tight and the RO water fittings work perfectly.
Remind me not to forget where I put my sunglasses… ![]()
![]()
2 Likes
Just make sure to purge with carrier gas for at least 15min before turning up the oven past ambient.
https://www.restek.com/catalog/view/49238
very convincing ![]()
1 Like
Even better, cheaper than the one I was looking at!
Being as this has not been run in a while and I am unsure of its condition. I’m wondering if I should proactively perform maintenance on the inlet before the first run. What are your thoughts?
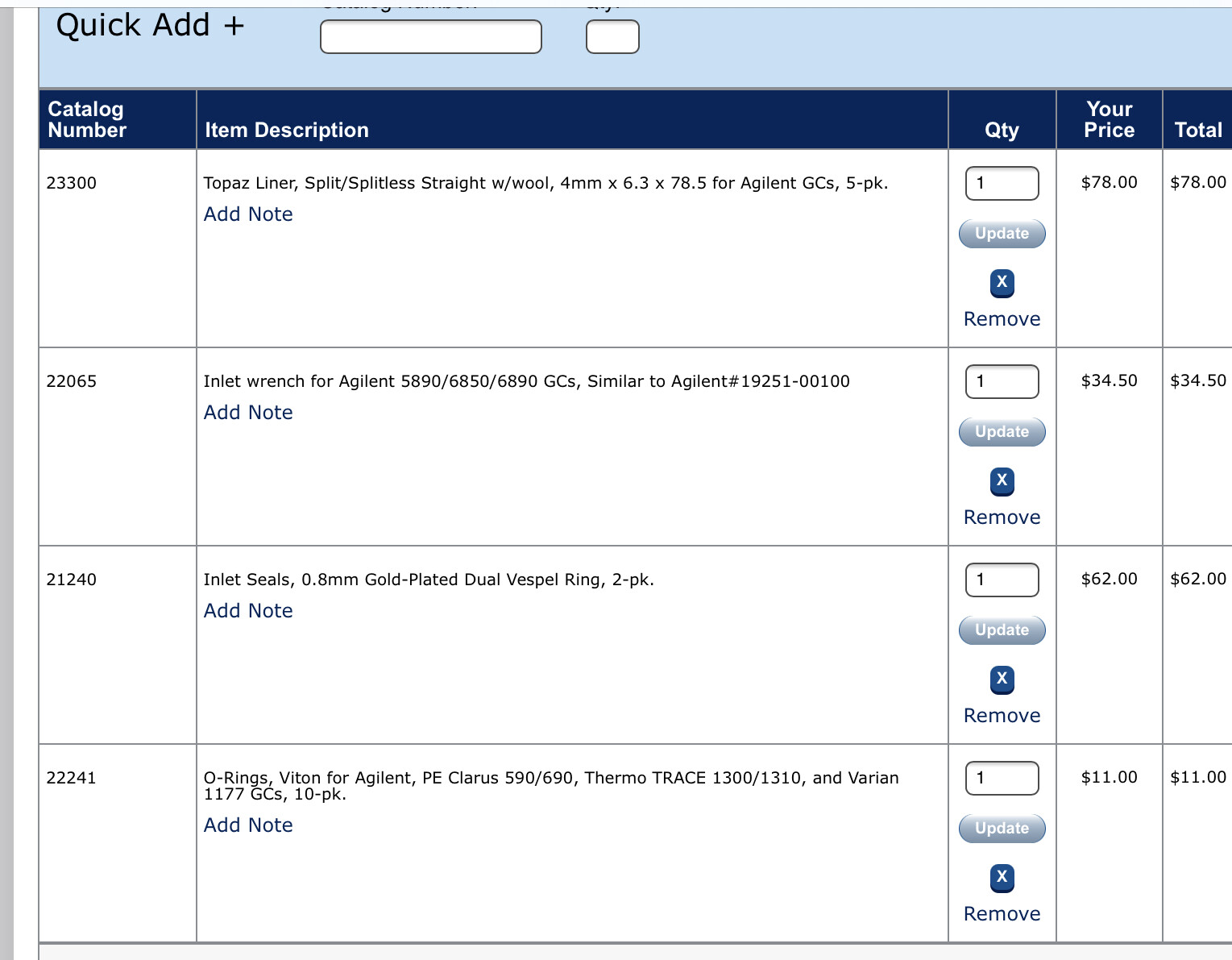
Do you already have septa?
Yes I would replace just about everything, though you may check the gold seal and clean it if it is not too far gone. Septa, inlet (liner), o-ring, definitely replace. Also check the split vent trap, depending on where this GC was used before it may be done for as well.
1 Like
Had something similar happen.
When we were moving labs one of my techs hooked up a GC with the hydrogen going into the inlet/carrier gas line. When the GC was running we had a small amount of unburned H2 going to the split but the rest was being burned by the FID. The big problem is our standby mode which is run 24/7 flows carrier gas through the column with the FID off so lots of H2 entering the lab.
We noticed a couple days into re-validating methods as all the retention times had shifted. Was not my first investigation but eventually I saw where the H2 was running, my heart dropped. Shut the tank, got everyone out and let the fume hoods run for a while, thank god nothing bad happened.
Our combustible gas monitor never picked up any H2 which was weird though it is across the room. At the end of the day probably not a lot of H2 was escaping or collecting, but still.
I imagine that would be the flow restrictor doing it’s job…
Yes I have septa and ferrules for 0.25 which match my columns ![]()
I’ll have to check tomorrow if I can get to the split vent trap. Might have to move things. I could not find specific instructions or a replacement part for 5890. Is it possible this was an addition to the 6890 and later? If you know the restek replacement part I’d be grateful
What in the holy hell is this?
2 Likes
Does this mean I can run a single test for both?
Not sure what exactly you’re looking for but you might look here:
rk125.pdf (971.7 KB)
1 Like
That document is quite helpful, and it does appear its a new model feature:

Well, with that being the case I think I’ll place the order and hope maybe to be testing by Wednesday
1 Like
Glad to be of help, can’t wait to see it up and running
1 Like
I’ve been trying to get my notes together on what the process is going to look like start to finish and understand where the gaps are that I need to resolve.
@Chaboes as you have the closest GC to mine that i am aware of, particularly appreciate your input.
I intend to derivitize with MSTFA following the general guidance laid out by @srihugh1 in his videos.
Opening and storing of Restek Stadandards:
- crack open bottle and transfer via disposable pipette to 2ml screw cap or other suitable storage medium
- store in freezer
Preparation of internal standard solution:
- 264mg methyl sterate
- 1000ml acetone
Creating a working standard:
- 100uL of standard is added to 2ml vial
- standards in methanol are evaporated and replaced with equal volume acetone
Creating an internal calibration standard:
- combine *working standard in equal volume to internal standard solution
- i.e: 100uL working standard, 100uL internal standard solution
Derivitizing the calibration standard:
- take 20uL of *internal calibration standard (clean syringe with acetone)
- take 20uL of MSFTA (clean syringe with methanol, then acetone)
Derivatization should per performed on cannabinoids with acidic forms, THCa, CBDa, CBGa, etc.
The derivitized standard and the internal calibration standard work to create an internal standard for testing acid and decarboxylated forms.
I am unclear, it is my estimation, that I will need to create and run each of these standards separately for calibration. It is unclear to me, what other steps may be necessary to insure an accurate quantification
Sample preparation:
- 100mg of sample is combined with 40ml internal standard solution
- sample is allowed to incubate (50-60c) for extraction
- 20ul of sample is taken and combined with 20ul of MSTFA
Questions about this process. Is the 40ml/100mg volume necessary?
Running the sample:
Inject 1ul of derivitized sample via 10ul syringe into injection port
I am unsure what the exact run parameters should be for the 35mx sil column at this time
From what I can gather from the Agilent training videos I could find, i believe I combine all the individual internal standards, identify them in the software and their known quantity. I guess that is then applied to a run to quantify the results?
Pieces I think I’m missing:
- run parameters for 35mx sil. Best as i can tell I should just copy this into Chemstation somehow: https://www.restek.com/chromatogram/view/GC_FF1248 or this one https://www.restek.com/chromatogram/view/GC_FS0549/5957-75-5 I’m not sure which to prefer as they have different oven settings; I’m inclined to follow the second one which shows d8/d9 being separated
- does the GC have it’s own airflow generator? I see it mentioned in the parameters but I’m not sure where the air source would be (and I did not see an air line on my machine)
1 Like
The expiration date listed on the standard is only applicable to the un-cracked ampule, once opened and transferred the shelf life is dependent on your own QC requirements. Typically I find about 6-8 months stored at -20°C.
It is important that you understand the dilution you are performing here. You are measuring a volume and a weight. Therefore, you either need to know the weight of 1000mL of acetone and add it to the weight of the methyl sterate to get total weight OR you need to add your methyl sterate to a 1000mL volumetric flask and bring the volume to 1000mL with acetone. Alternatively you could add 264mg of methyl sterate and bring to 1000g with acetone. These are all different dilutions (w/w), (w/v), (v/v) and the distinctions are important when designing the method.
Guessing you meant 100µL. Also what is the point of doing this (not trying to be rude just trying to understand)?
Again I’m guessing µL not mL here.
External standard (ESTD) will be at 1000µg/mL
Using at w/v for your internal standard solution:
(Bring up to 1000mL in volumetric)
264mg/1000mL= 0.264mg/mL or 264µg/mL
100µL of ESTD = 100µg to vial
100µL of ISTD = 26.4µg to vial
200µL total in vial.
Internal Calibration Standard Concentrations
(26.4µg/200µL = 0.132µg/µL [ISTD]) (100µg/200µL= 0.5µg/µL [ESTD])
20µL * 0.132µg/µL [ISTD] = 2.64µg to Derivatization Vial
20µL * 0.5µg/µL [ESTD] = 10µg ESTD to Derivatization Vial
At this point I would suggest drying the sample down before adding the derivatization agent, helps with calculations later on.
Let’s say we do not and simply add the 20µL of MSFTA
10µg/40µL = 0.25µg/µL of ESTD or 250ng/µL
2.64µg/40µL = 0.066µg/mL of ISTD or 66ng/µL
These are both relatively okay analyte loads to hit your column, typically I would try to stay under 150ng of analyte on injection. Do not forget that if you are injection with a split the actual load on the column will be drastically decreased based off the split ratio.
How are you planning to control this in a real world sample?
Derivatization will take place on all the cannabinoids, remember we are targeting active (protic) hydrogens and almost all cannabinoids have at least a hydroxyl group. This means if the only standard derivatizations you are doing are with the acidics you are not going to have the right retention time for the derivatized decarboxylated forms.
You will need at least a 5 point calibration curve for each cannabinoid to be quantified.
(e.g. 10ng/µL, 25ng/µL, 50ng/µL, 75ng/µL, 100ng/µL)
This workflow is off. The incubation needs to take place with the derivatization agent. You do not need heat for the extraction. Extract at room temperature, take a small sample and then derivatize and incubate.
Remember we want to keep the analyte load <150ng on column.
You have a okay foundation for this method but I think you need more research in order to understand the serial dilutions that are needed as well as a proper workflow. I’m confused at some of the volumes you are using and how you are preparing your standards.
5 Likes
Let me preface this by saying all this is derived from breaking down information I was aware of and available to me, in an attempt to put together the blue print for a process; absent any formal process to build from.
Yes, my apologies, when I transferred my notes I typed them out incorrectly and didn’t double check. 100ul of standard and uL where you suspect.
As to the point, my understanding of why is methanol deactivates the MSFTA, and needs to be removed; acetone doesn’t interact and is used in place.
Thank you for laying this out as you have, that helps put things into perspective. I suspect the figures (which translates to 1 gram per 1 gallon) are done for ease of preparation rather than towards making a standard as you have outlined here; i much prefer, seeing now how you have laid it out, to make equal the volume of the ISTD and the ETSD. I assume this would also roughly translate to the same sized peak?
Am I to understand, you are inferring the ESTD should come down and equalize to approximately 75ng/uL each ESTD and ITSD;
I’m not entirely sure the effect this statement implies. Does this infer the higher sample load might be less of an issue because the column will only take part of the injection anyway?
My expectation, is to have performed derivitization on the non-acid forms to measure their retention time, and then measure the retention time of the underivitized forms; as I understand it, in a derivitized sample this becomes the measurement point for the acid form and the derivitized for the already decaroboxylated forms.
My expectation, based on the above understanding, is that universally derivitizing should produce consistent results and I would think i would derivitize every sample for processing. Again, this is only my expectation based on my crude understanding of the process.
Am I correct to understand this means, as I prepare the working standards, the injection load should be at these levels (assuming equal amounts of ISTD and ESTD; so 10ng:10ng?) as part of the calibration curve? Are any other points needed to create an accurate calibration?
I’ll have to double check, again having no experience, I took notes; incubation happened at sample prep and derivitization then sample was essentially immediate.
Starting from no foundation, hopefully explains some of it ![]()
5 Likes
You’re right, good point.
Personally I would try to limit the changes you make to the standard as it will introduce variability. You could instead create a separate IS solution and add that as well as your ESTD to a vial, dry down, and then reconstitute in ACN or Acetone + MSFTA and perform your derivatization in that vial. That way you avoid one intermediary step.
No real need to make sure the peaks are equal, having a IS peak roughly 1/4 of your ESTD peak is not an issue. All that matters is you know what the response is supposed to be, and how they two relate to each other.
Lets say we have a split ratio of 1:20, meaning 1 part to every 20 of the 1µL injection gets put on the column. So if our sample pre injection is at 250ng/µL we would be injection roughly 12.5ng onto the column. This is simplified but since all your samples are being run with the same split ratio including your standards, all that is important is knowing your sample and standard concentrations, the split just helps to load less sample on the column.
Gotcha, just wanted to ensure you were derivatizing all cannabinoids.
Actually you will want to keep the IS at a constant concentration while changing the amount of ESTDs when building you calibration curve. Chemstation will want a constant IS concentration and will look to see how that relates to changes in ESTD throughout the curve.
(e.g.
Cal Point 1: 10ng/µL CBDa and 10ng/µL IS
Cal Point 2: 25ng/µL CBDa and 10ng/µL IS…
and so on)
3 Likes
If I wasn’t clear, my intention would be to never contaminate the standard, and should always be transferred via disposable pipette from the standard vial to a clean vial; and the standard vial is to be kept in the freezer.
Yes, in hindsight it was a short sighted question ![]() . This makes a lot of sense.
. This makes a lot of sense.
Would it be accurate to summarize as follows?
- the 264mg ITSD figure doesn’t need to be specific and should be formulated for simplicity and accuracy.
- all standards, including the terpenes, should be derivitized at the 5 calibration points with an equal amount of itsd per calibration point. (As best as possible anyway).
Questions:
- Am I correct in understanding I only need to do this this process of calibrating with standards when my internal standard solution changes (presuming I am imperfect in measuring/formulating)
- Can I run a cannabinoid and terpene test at the same time on the 35Sil MS column?
- Anything obvious I’m missing, or any tricks that make these processes easier? I’m assuming the calibration will be the biggest thing to get correct for accurate results