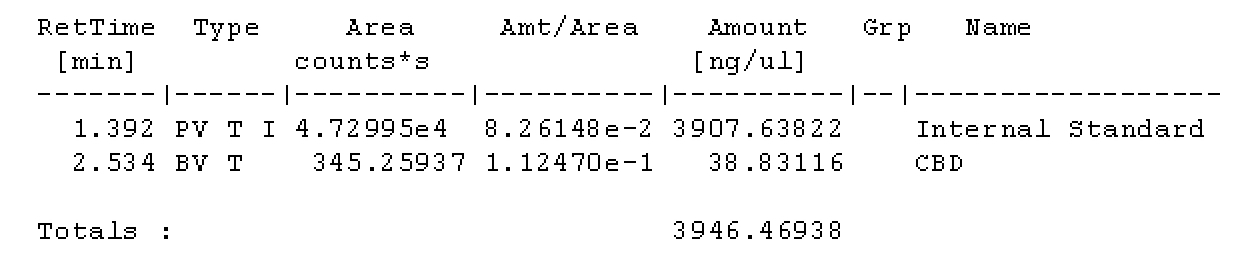
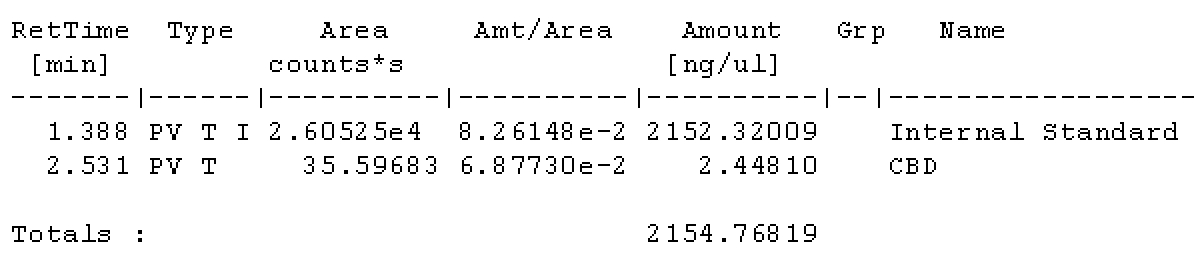
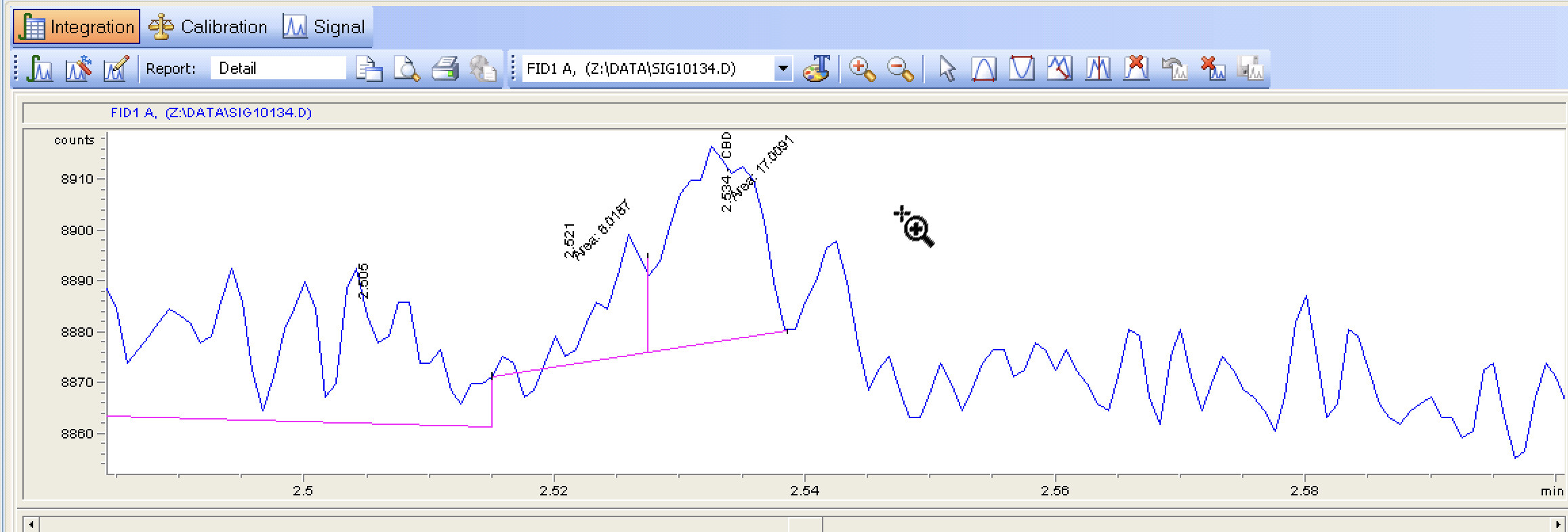
I wanted to make sure this was the case. Please by all means check my math because I could have overlooked it!
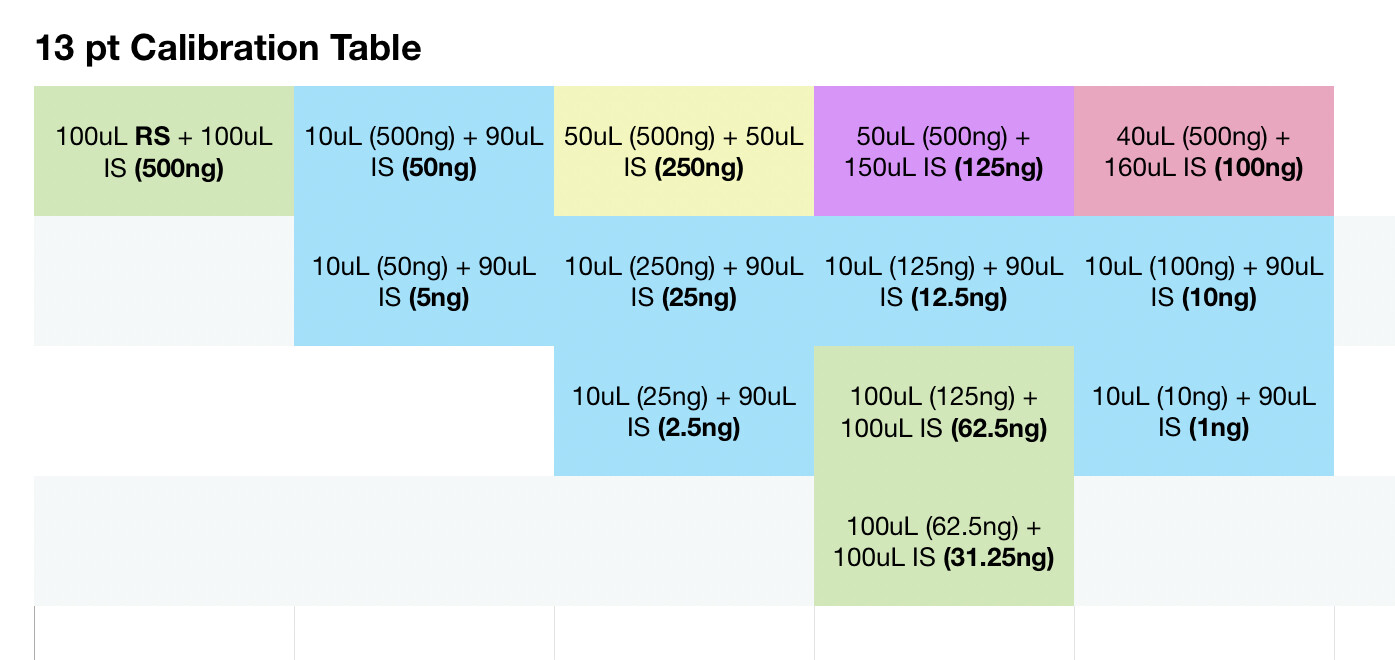
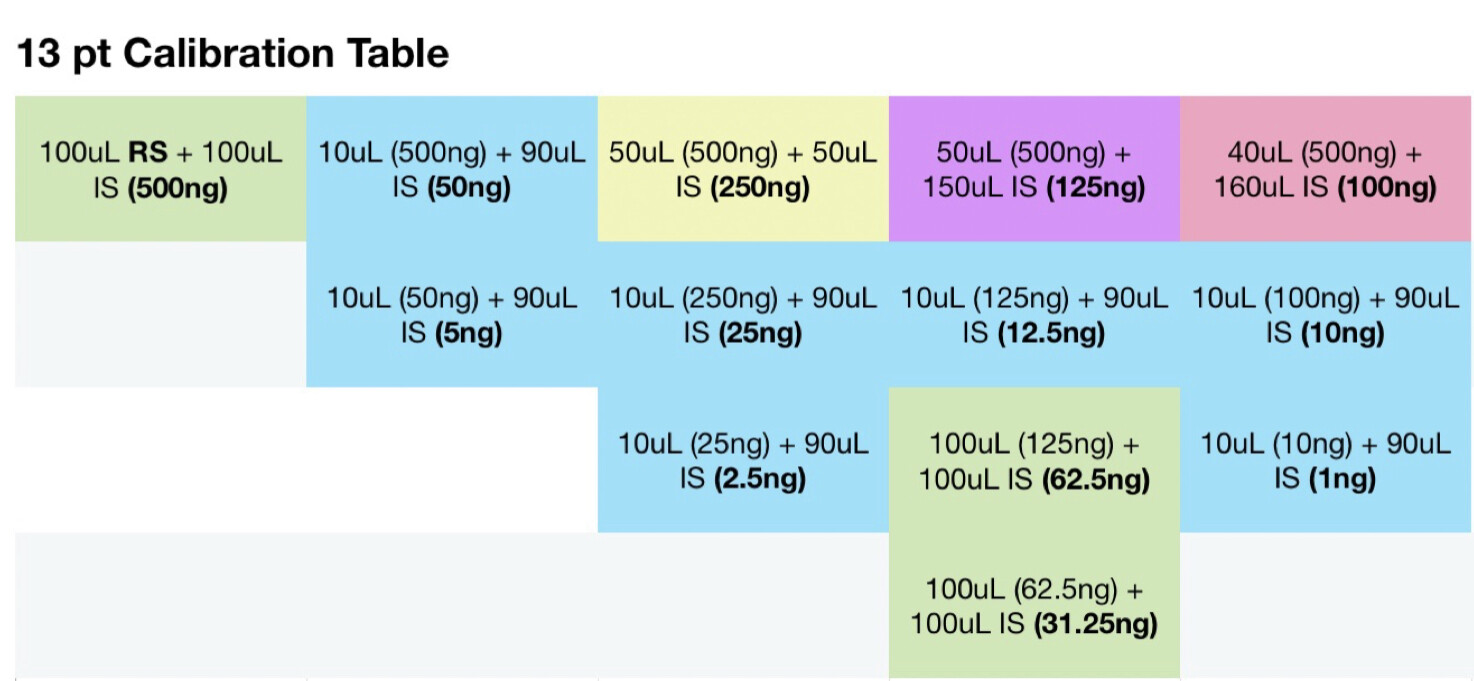
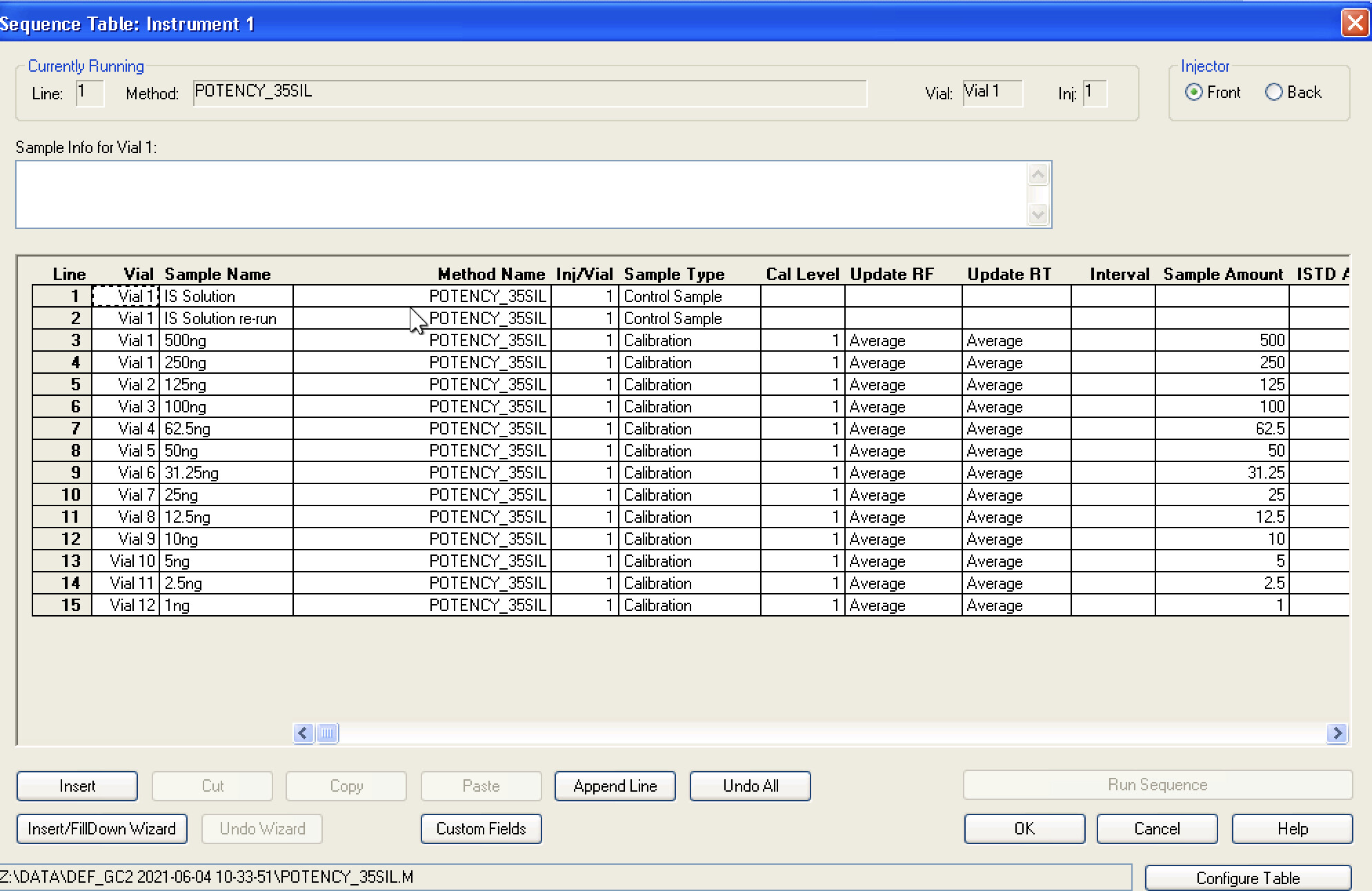
To one vial add 100 uL restek standard, allow to evaporate. Add 200 uL IS solution to this vial and seal. Then perform the following:
With each vial filled with IS solution, everything should be at the same concentration level of IS per uL. Am I mistaken? If I take 10uL of solution from the master vial above, the amount of IS should be the same, per uL as the vials with only IS solution. Maybe my brains fried, it very well could be.
I don’t mind being wrong, if I’m doing something stupid let me know!
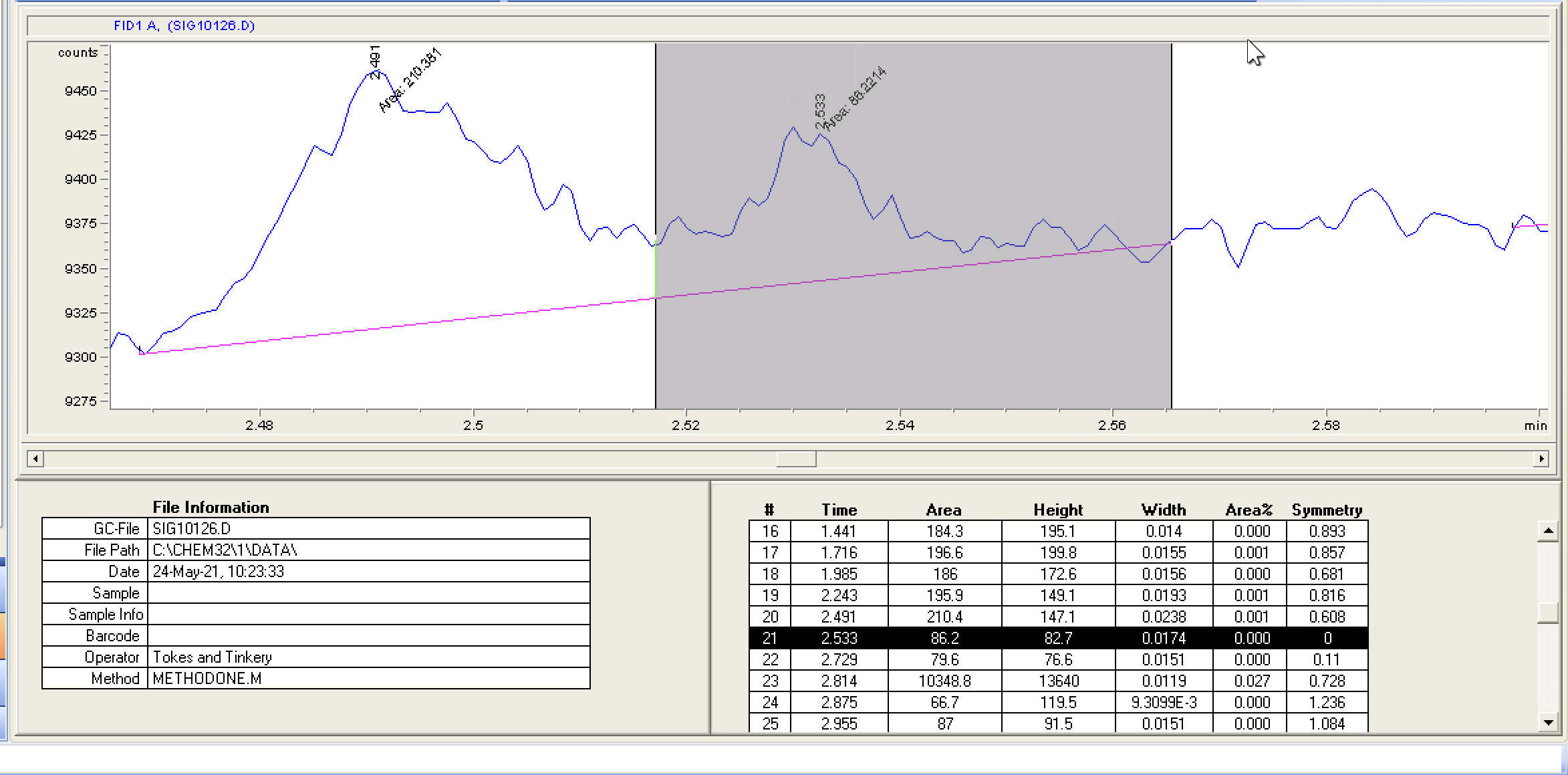
Side question, is there any way I can change my view to match yours? I’m really at a loss at how the baselines compare with the differences in signal measurements.
I trust my brain could be fried, maybe double check my math?
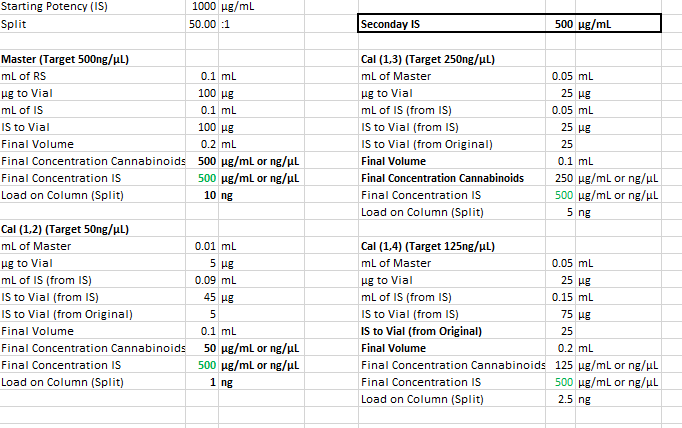
Internal standard prep:
- 1.059g methyl stearate
A. 1059mg (1059000000ng)
- 749.1g acetone
A. 954.75ml (954750ul)
1.10919 mg per ml
11mcg per ul
1109.19ng per ul
With a 50:1 split that should be 22ng on column if I’m not mistaken?
For sample prep, if I aimed for 100mg compound to 40ml IS solution, assuming 100% potency that would be 2500ng per uL and at a 50:1 split should be 50ng on column. If thats accurate, i probably at the very least need a calibration point higher, would you agree? Maybe another point at 100uL standard evaporated and added to 100uL IS for a 1000ng/ul calibration point, and 20ng on column. Is that close enough or should there be calibration point nearer the 100% purity mark?
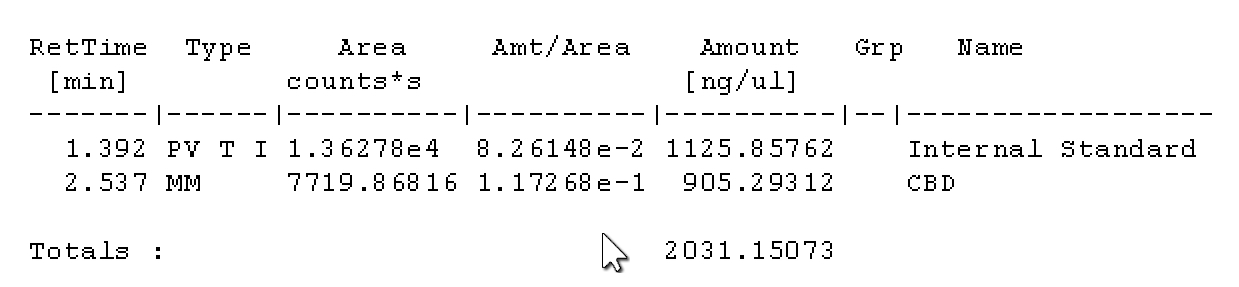
Seems my calibration table favors the lower end, i guess it’ll be interesting to see exactly how low the LOD will end up being. It seems kinda neat to me, this sample of flower I ran in the above looks like it has trace d8.
Maybe I’m overthinking it