I appreciate all the input. Thanks again to both you @Chaboes and @cassin as well.
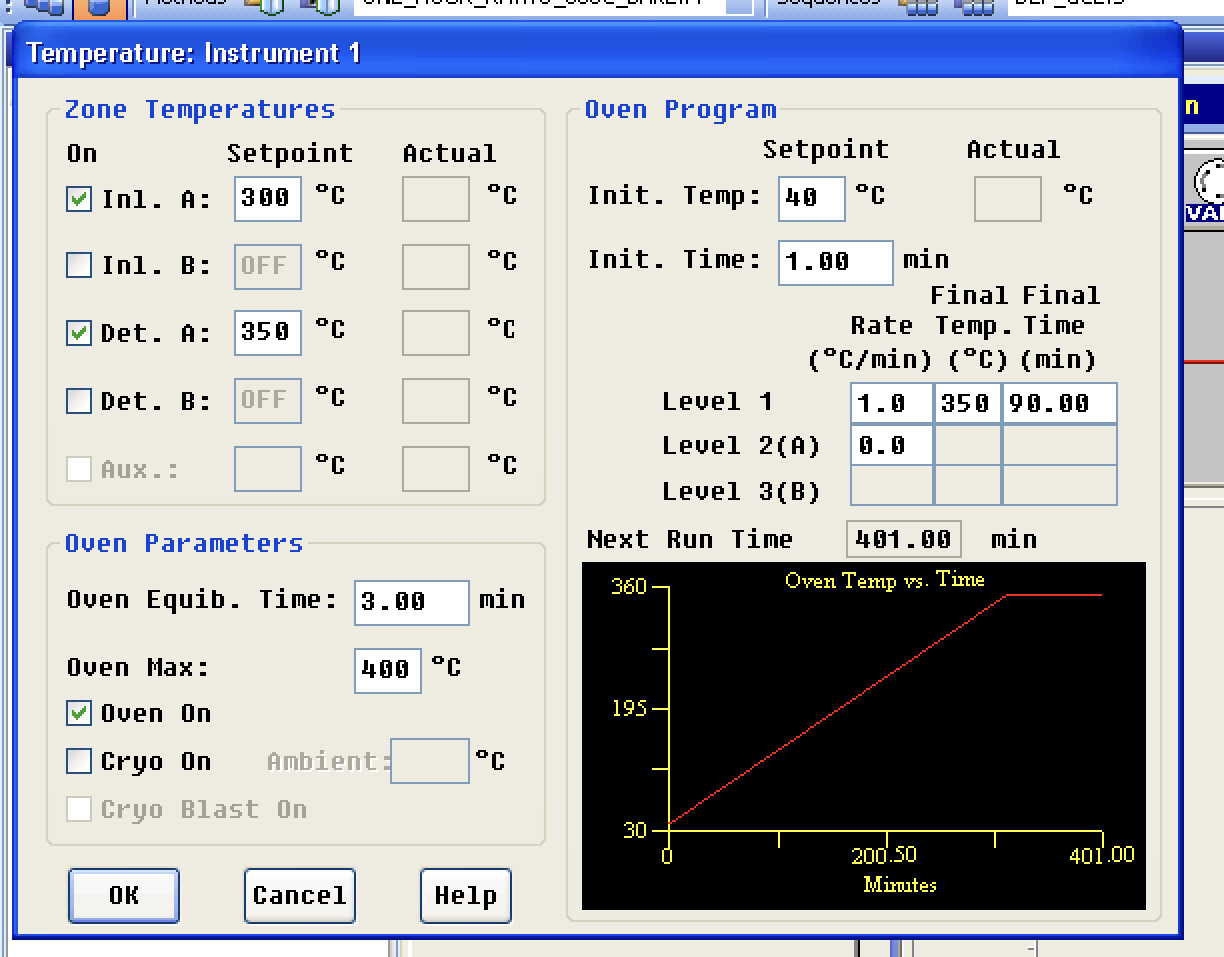
I’ve got the 35sil cut cleaned and back in place. I had to readjust the airflow settings to get back up to the original numbers. Additionally I have changed the split vent flow to 50ml/min or apx 20:1 split.
Originally I was following one that a split vent of 50:1 but I don’t think I can hit those numbers with the valves I have. I’m not entirely certain here.
Currently flowing hydrogen through the Column and then will begin to heat and rebake the column for a couple hours.
The next big unknown hurdle is doing the calibration curves. I’m moderately terrified of ruining my standards
Just take an aliquot from your standard with a new pipette and put it in a sealed vial. Then you have your original still available for next time. Keep the original in your -80C and call it good.
I usually do like a 100uL aliquot with 900uL of whatever chromatography grade stuff I have available.
If you don’t have clean pipette tips… or calibrated pipettes to do this… you should get some. There’s super super cheap ones for like 75$ - they will be accurate enough to get you started, but investing in better or even a set of Hamilton syringes is where its at for this kind of stuff.
I’ve got the pipettes, tips and 2ml vials with septa’s ready to go. My plans was to pipette out of the ampule once broken and into the screw top vial seal, label and then pop back into the freezer, only -20C here unless I put it in a cold trap bath!
Should I rinse the pipette tips with a solvent flush? What about the vials? Is it necessary?
I think the biggest upcoming challenge, or unknown is getting the calibrated solutions done properly and put into the machine.
How often do you re-run your standards on your machines? Daily? Weekly? Monthly? Otherwise?
No. You can use them right out of the package most times, no issues. Use them once and discard them unless you are aliquoting the same thing over and over. Should be able to fit your whole standard into a single 1mL vial - unless you got the big standards, nothing wrong with putting it into multiple vials if you want either.
I don’t rinse the vials with anything - but I also always make sure everything comes sealed and is laboratory grade. Only time I rinse and clean everything is for heavy metals testing - otherwise I use it right out of the package.
Calibration standards are not hard. Just start with a 10 by dilution - (assuming 1000uL/mL):
100uL into 900uL now you have a 100uL/mL solution
Take 100uL of that into 900uL and you have a 10uL/mL solution
Take 100uL of that into 900uL and you have a 1uL/mL solution
That gives you three points - if you want 5 points - do two midpoints between 100 and 10 and 10 and 1. By doing 50uL into 950uL from the previous dilution.
Five points should be enough - you can always make things more potent up the original concentration of the standard. There’s some room for fenagling here I’ve been told, but I always work with liquid injections not headspace stuff.
I run standards every single day I’m running samples to verify that my calibration still looks good. And then I run a single standard every 10-20 samples (based on if I see any drift or not that I need to monitor for).
I do a full calibration anytime I see more than a 2% RSD drift or once a week whichever comes first. And I run a full calibration anytime I do any maintenance at all - septum change, calibrate, new column definitely calibrate, change out a tank of gas, calibrate. You get the idea.
-20C will be okay - but I have seen degradation even in the freezer after 6-9 months… so just monitor for it, you’ll know right away if you are getting protonation or methylation or dropping your acid groups because there will be unexpected new peaks and peak area changes.
I monitor for everything in a trend log - that way I can see when drift is happening. It helps me to know when I “need” maintenance instead of just doing it because someone thinks I need to as well.
I’m going to run a series of injectionless blanks to see how they compare next. I think i may also try recutting and installing the column to see if the baseline improves. It was far better on the other column and maybe its due to installation?
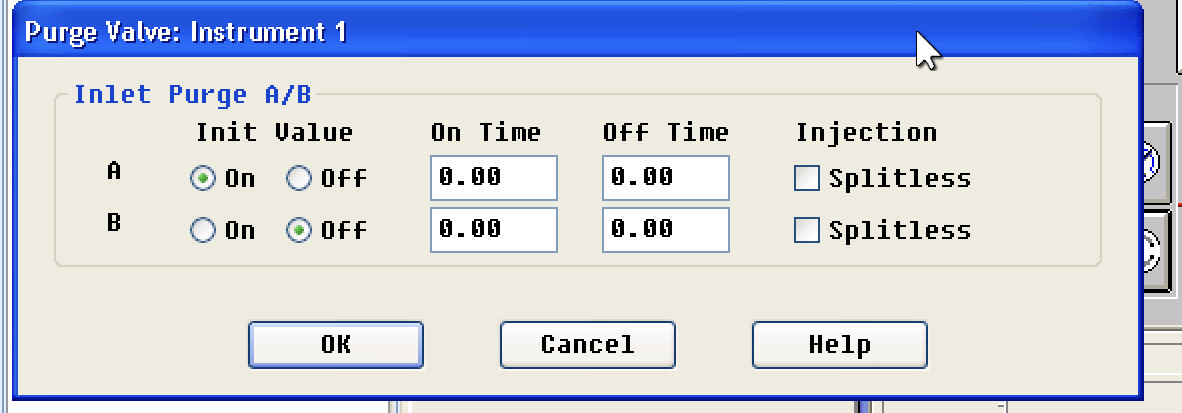
@Cassin or @Chaboes, do you have a setting for purge on/off? Curious what your timings are as mine are 0, not sure what it should be if anything
So you will probably need to set a purge time. When running spitless you are assuming the entire sample is to be loaded on the column but eventually you want to get rid of anything left in the inlet to clear it out. If you do not set a purge then leftover nonvolatile species will be left in your inlet possibly coming off very slowly over time (next injections) as well reducing inlet liner and column life. With complex matrices like cannabis and cannabis products you definitely will either need to purge or run split. It is pretty easy to set this timing, here is a calculator
I completely missed that field at the bottom of that page, thank you for pointing that out!
I setup for 0.5 to 0.8 and will do a few blank runs to see how it looks. I’ve been running back to back blank runs after changing out the septa, clean & cutting the column. First run was very spikey, each run after is less so, or somewhat randomly so at this point.
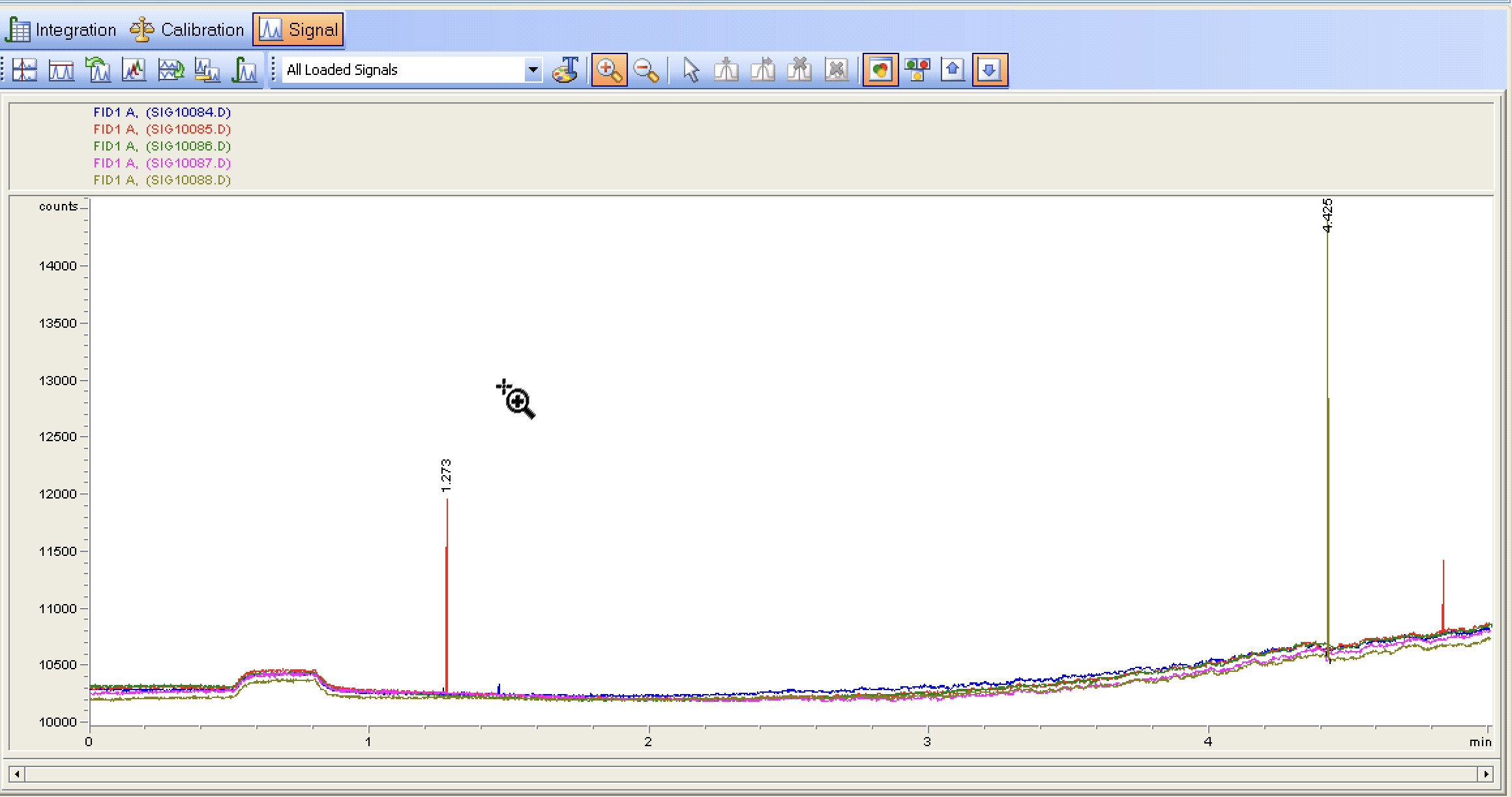
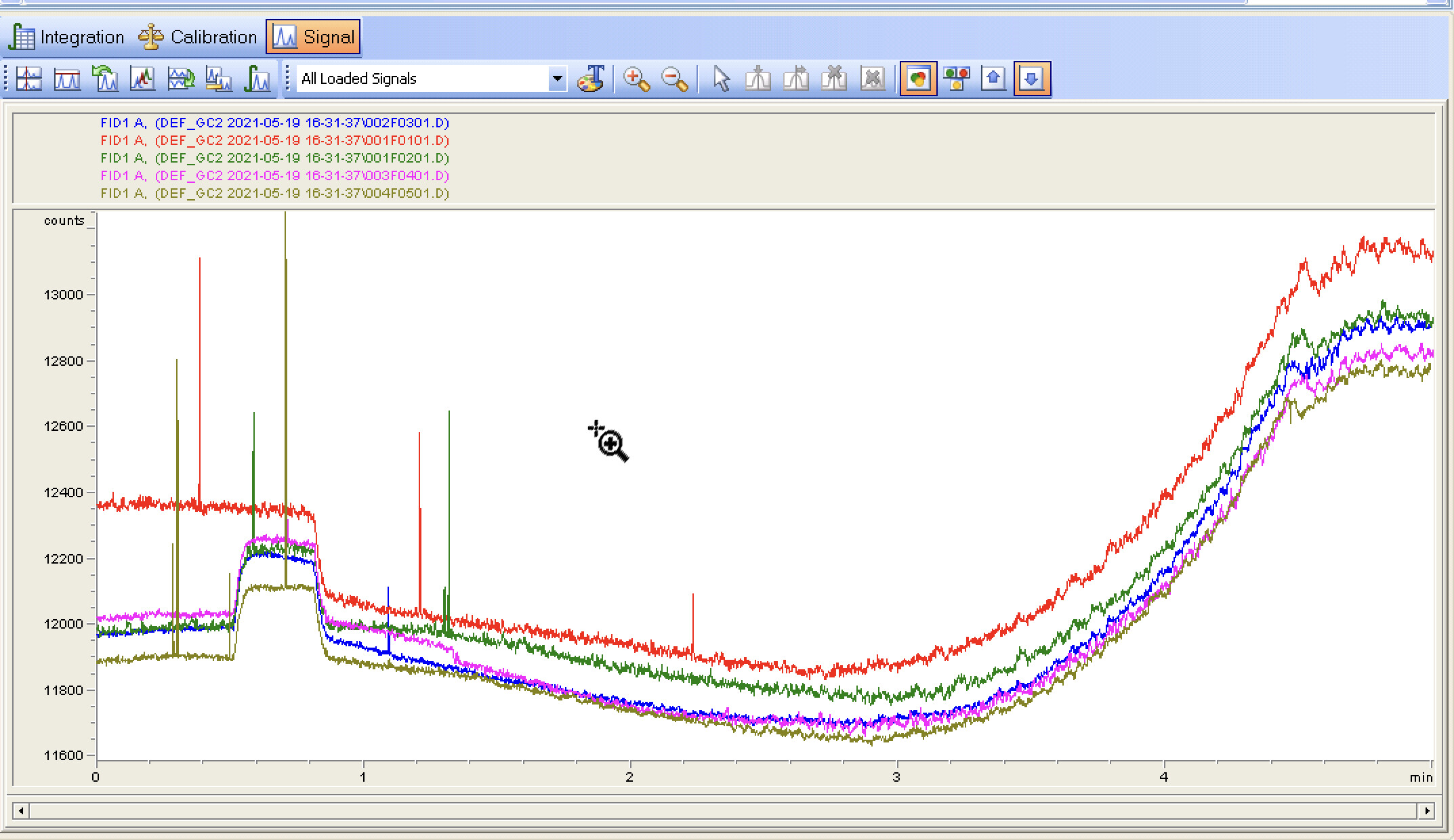
The last 5 runs with the purge valve settings on. A few random spikes and the baseline signal continues to go lower (I assume as some form of conditioning on the column?)
I decided to check the flow rates and somewhat to my surprise they are off. I hadnt considered the flow rates measured at room temp would change as the column was heated. I’ve rechecked all the settings, this time I am back to a split vent flow of 125ml/min or 50:1 split. Going to run some baselines now
Been conditioning the column off and on all day, currently just cycling through. Decided to spice it up a bit and run a sample of the crude and see what comes of it.
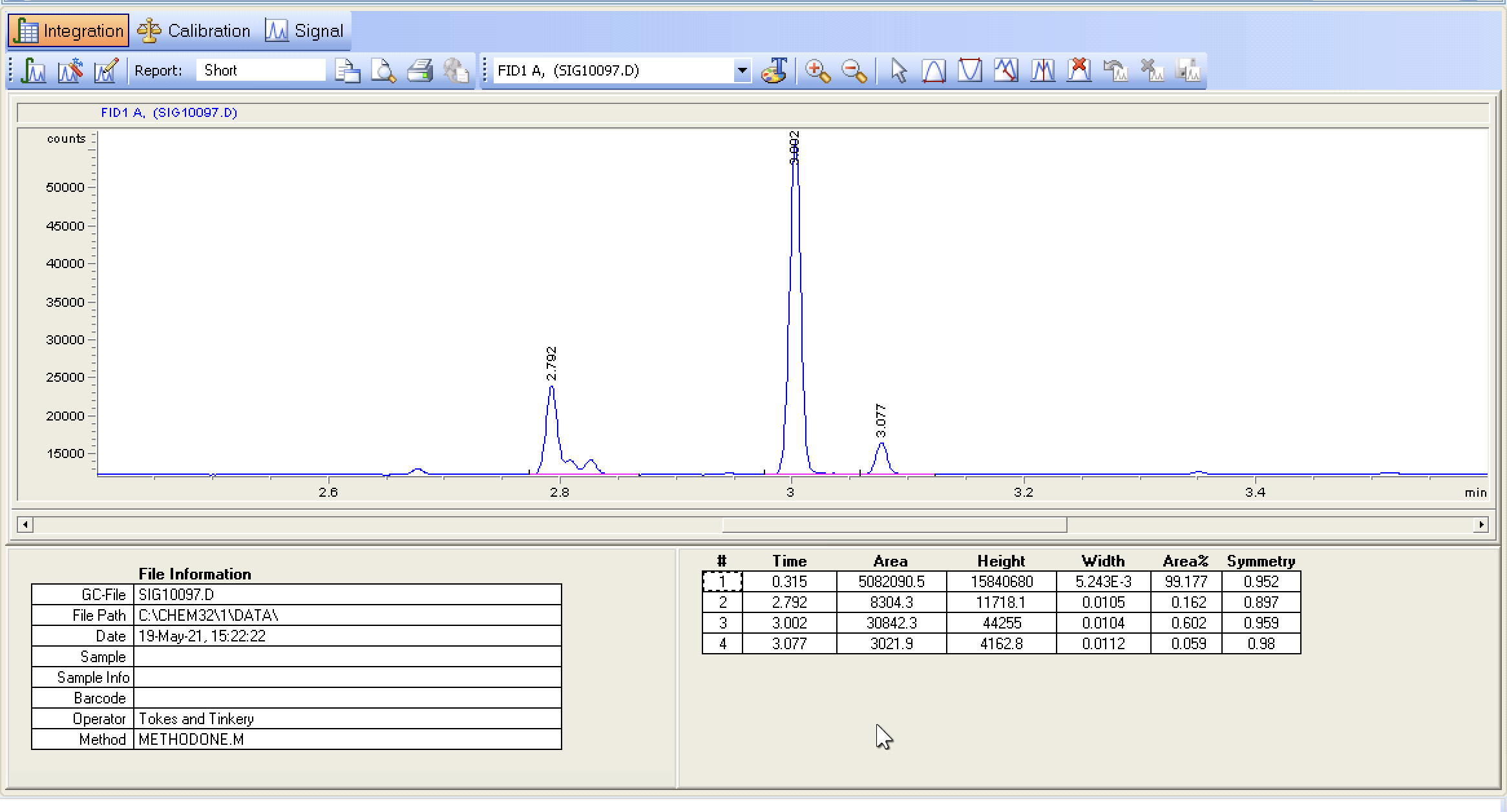
The separation looks pretty decent, still getting some inconsistencies on the baseline/blank runs. I wonder how much, if anything, will change with the new column.
My lack of experience in Chemstation is certainly evident as I try and make sense of the data for presentation. Understanding the integration settings and getting those right is still elusive to me at this time.
Overall, very happy with the progress so far. Still intimidated by the calibrations stuff; both process for prep and run; as well as doing it on the Chemstation side.
I notice the baseline continues to lower run after run; @Chaboes@Cassin should I expect this to settle eventually or is this part of the integration process, having to compensate for these?
Not sure what to make of the random peaks, possibly compounds eluting? Could also be related to the purge valve going off? Not sure at this time.
I think I’d like to order some proper microliter syringes; I can see the damage the sharp edge is doing to the septa. I think a proper conical tip syringe is in order. @Cassin@Chaboes and anyone else watching; would you have a specific product recommendation to order?
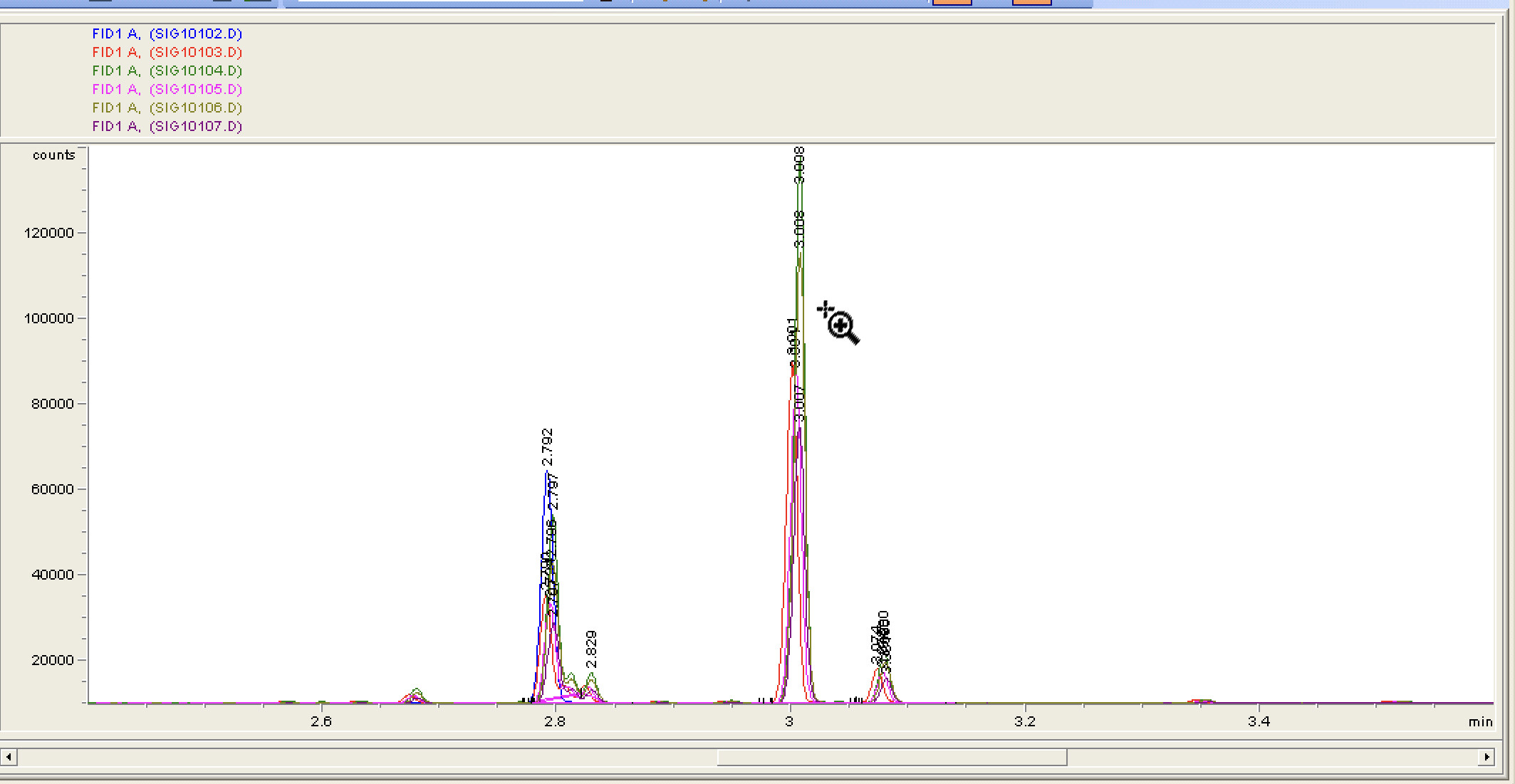
After letting it run continuously, this is what we’ve got (this screen shot shows the window where we mostly expect to see stuff)
I think a few runs with the same sample to test retention time may be next. The baseline seems to keep going down, I wonder where it will eventually bottom out. I suspect if the retention time tests look good I’ll have no choice but to crack open the standards
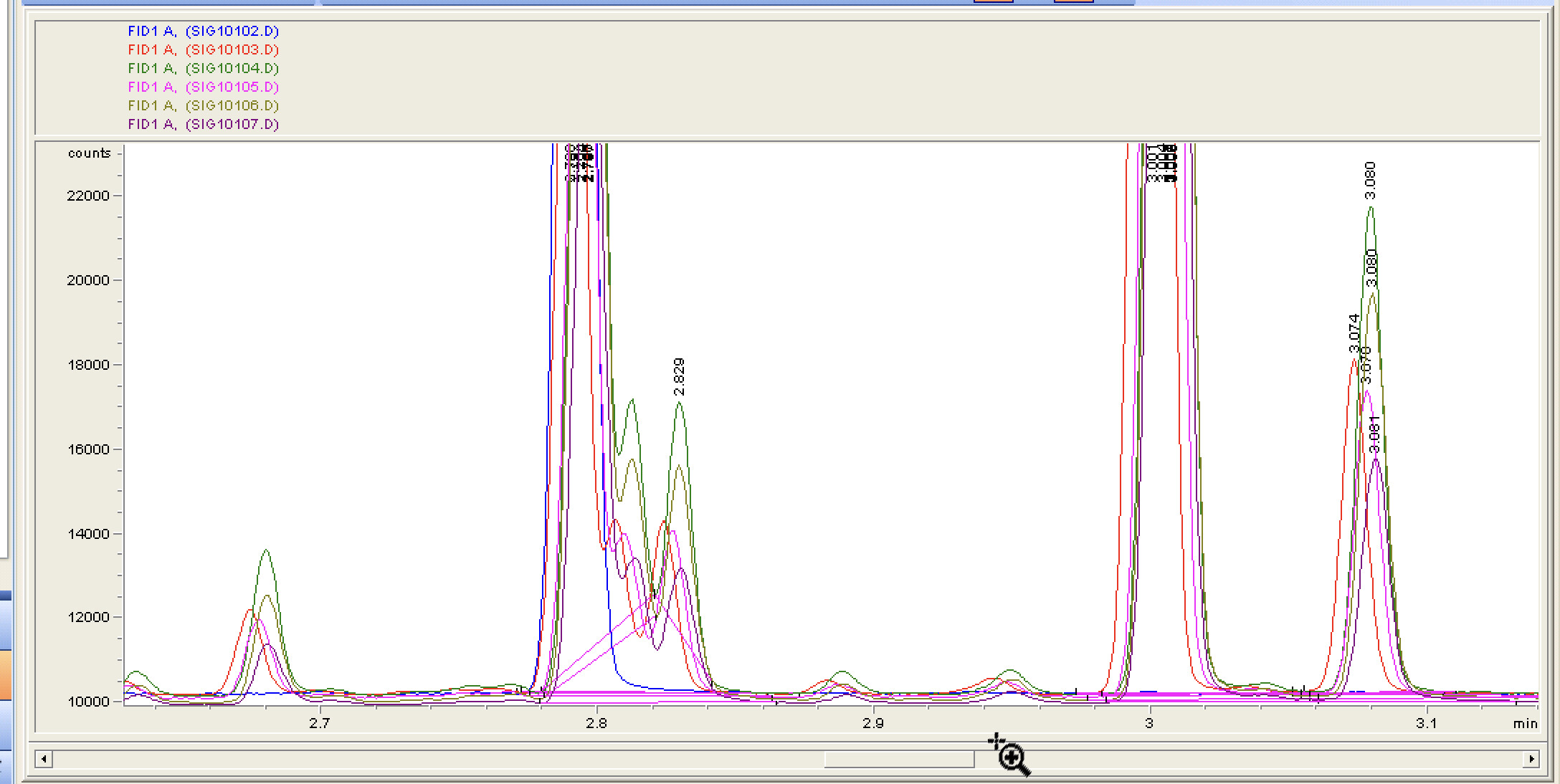
Here we go, I did not adjust these to line up so all in all, my injection technique is at least mostly consistent? (6 manual runs)
Doubt we would be able to identify what is following the CBD peak, its probably non-cannabinoid plant extract from the crude.
Digging deep into our old data I was only able to find one run of a d8 which was a “skittle” that was received at an Expo. It did seem to have peak very close to where CBD would elute for us which was interesting, but it was before. Who knows, in total three unknown peaks but probably related to the candy matrix.
100% - this baseline looks amazing. It will get better for a long while as the column keeps conditioning - and then at some point it will start getting messy because your samples will have caused part of the column to get gunky and you’ll start seeing weird stuff in your blanks. The S/N looks awesome though.
And you might have some sample prep issues to handle - sounds like its time to break open those standards and get cracking on calibration. Then you can worry about sample preparation to make sure you get consistent results there.
Are you talking about the random spikes that have almost no width? If so, this could come from a couple of different sources.
Valve switching events are a possible cause, but I don’t think this is the case since the retention times of the spikes are totally random.
When I see these spikes a dirty detector comes to mind. My first corrective action would be to clean the FID. During this process I would check the electrical connections.
You could also have particles exiting your column and hitting the detector. However, this is not very common when using bonded capillary columns.
Another source could be electrical noise, which can be hard to identify and correct.
I’m a little worried with the 50:1 split ratio the material load on the column is likely to be quite small based on conversations with @Chaboes in the past.
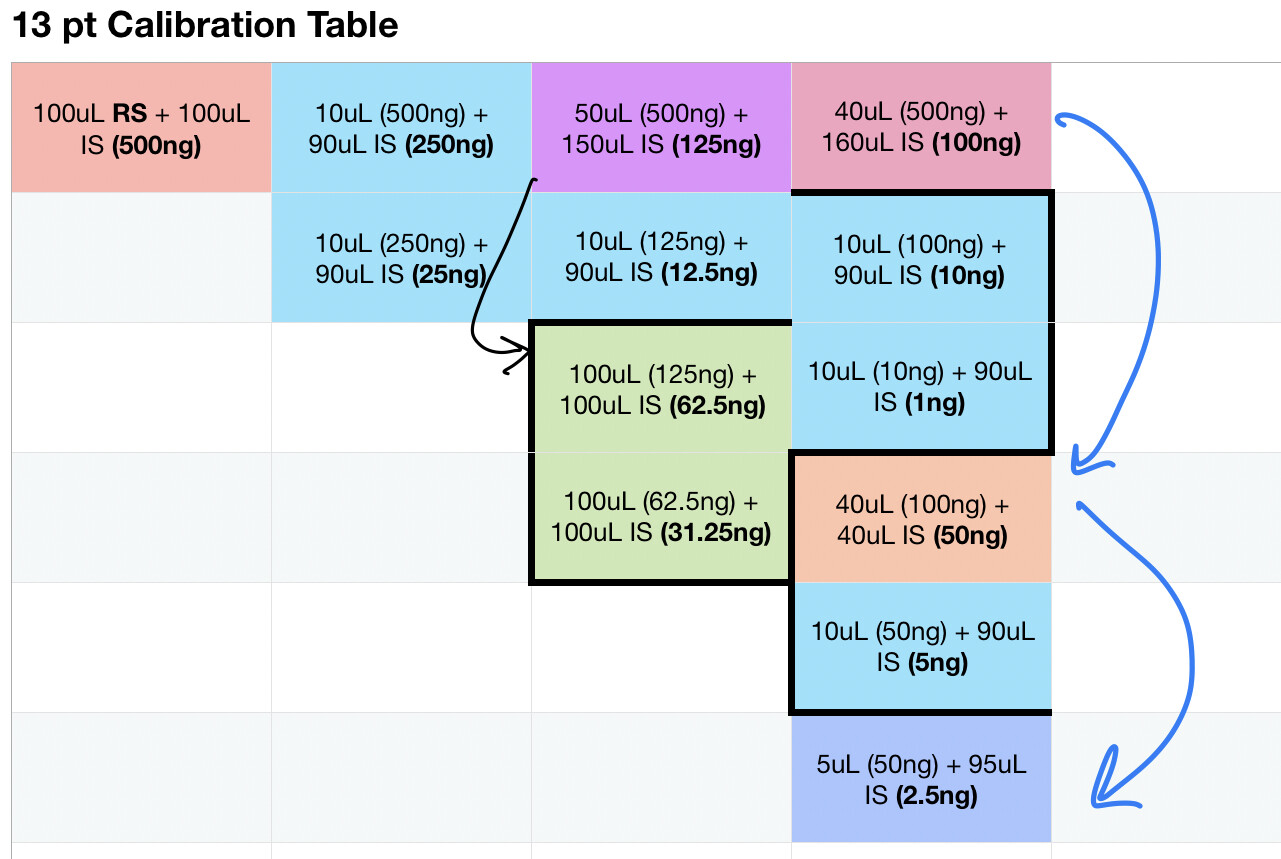
Nothing too special about the above, mostly designed to be conscientious of the amount of restek standard is needed to build the calibration set. I think I will plan to put 1:1 of the cannabinoids standards into the top left jar, evaporate off the solvent then add 200uL of internal standard solution which is the basis for the rest. I should note that the bold numbers in the graph represent the anticipated compound load per 1uL injection
If this calibration protocol holds up, it should be good for 75 complete recalibration and 100 on most points on the curve
If its of interest here’s the table I built the graph from including the approximate number of tests you should expect to get from it.
100uL RS + 100uL IS (500ng - 100 tests)
A. 40uL of original (1) + 160uL IS (100ng - 145 tests)
a. 10uL of 1.A + 90uL IS (10ng - 90 tests)
1. 10uL of 1.A.a + 90uL IS (1ng - 100 tests)
b. 40uL of 1.A + 40uL IS (50ng - 65 tests)
1. 10uL of 1.A.b + 90uL IS (5ng - 100 tests)
c. 5uL of 1.A.b + 95uL IS (2.5ng - 100 tests)
B. 10uL of original (1) + 90uL IS (250ng - 90 tests)
a. 10uL of 1.B + 90uL IS (25ng - 90 tests),
C. 50uL of original (1) + 150uL IS (125ng - 90 tests)
a. 10uL of 1.C + 90uL IS (12.5ng - 100 tests)
b. 100uL of 1.C + 100uL IS (62.5ng - 100 tests)
c. 100uL of 1.C.b + 100uL IS (31.25ng - 100 tests)
I intend to do 1g methyl stearate to 1000ml acetone for the internal standard solution targeting the same concentration as the standards themselves.
I’ll run 1uL injections with and without the purge valve on to see what it all looks like.
I might proof this out with CBD isolate (10mg + 10ml acetone) first…
I am a bit confused on your calibrations, everyone shows their work and thinks differently though so it very well could be lost in translation. The table in the graphic and the table in the post are not in the same order (column by column (L->R) as compared to top to bottom) but I think I figured it out.
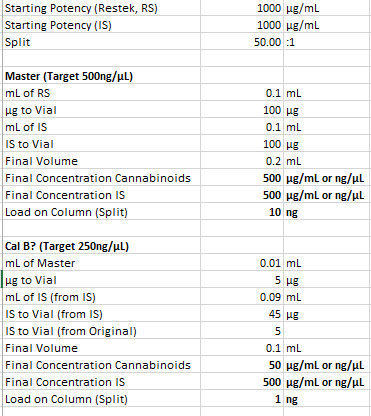
Creating a 250ng/µL calibrator from the master (original stock) would only require a 2x dilution since it is at 500ng/µL. What the math seems to show is a 10x dilution.
10µL * 500ng/µL = 5000ng or 5 µg
Final volume seems to be 0.1mL (10µL Master + 90µL IS)
5µg/0.1mL = 50ng/µL
I haven’t checked the other calibrators but it seems the IS concentration is staying constant which is impressive considering you are adding it both to the master stock and as an addition.
I may be wrong on the math but if this above check is correct you may need to redo some calculations.
You are correct, I think anything below 1ng on column is unnecessary. My lowest amount on column is for d9-THC which has about 0.12ng loaded. While this amount is detectable I do not use it as a calibration point, it was only used for LOD data while validating the method.
This is what 0.12ng on the column looks like. It is okay when running standard but this will become a lot more messy with matrix and is subject to large percent change in area with little changes in sample prep. You will want to stay a bit higher overall (depends entirely on the sample preparation and expected range)
In this case anything calibrated below ~10ng will be pushing is with a 50:1 split. But that is my machine/method not yours, you may have better sensitivity.