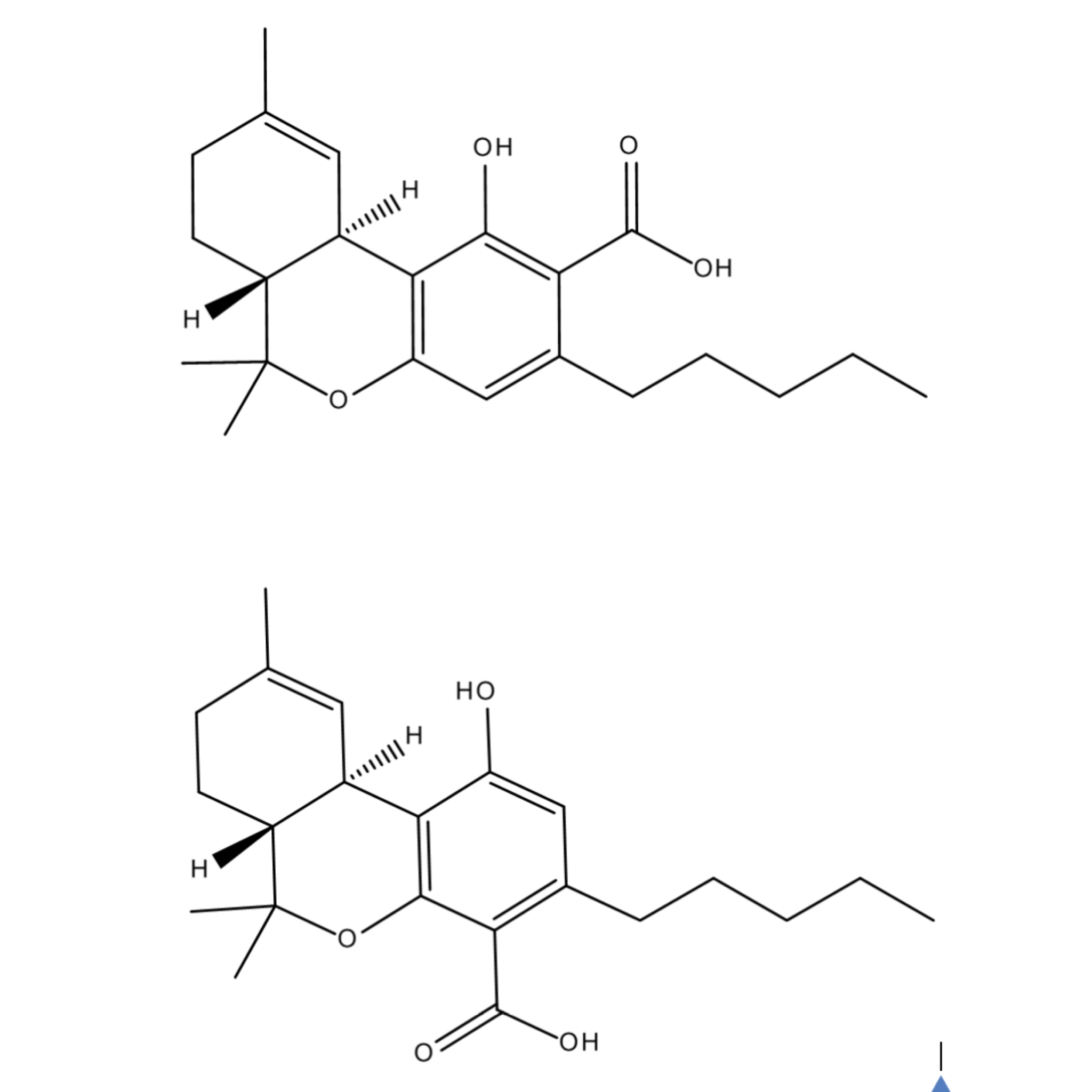
These are geometric isomers, right? Is there potentially a third or even fourth geometric isomer for THCa?
I see that the pentyl group could move around various places on the “ring”. The carboxylic acid group and the hydroxide could also move around that “ring”. Does the literature show otherwise?
Also, I know that stereoisomers don’t really have exactly similar properties. However, onced decarbed, would they still give the same THC molecule?
From what I understand through an interwebs search, both A and B can be decarbed into D9-THC. Meaning that you don’t even have to finish the salting method if THC is all you care about.
These are Positional Isomers because they only differ by the placement of the carboxylic acid functional group on the benzene ring, either at the 2 (THCA A) or 4 (THCA B) position.
In theory, there could be a 2, 4 dicarboxylated version of THC. THCA B is pretty rare in nature, so a doubly carboxylated THC, I imagine, would be even more so.
Think of the functional groups on the benzene ring as being pretty happy and well locked in the way are. Neither the pentyl tail, nor the hydroxide, will migrate. What can migrate is the position of the double bond in the (as we’re looking at the above pictures) upper left ring. The current position of the double bond is that of delta 9 THCA’s.
Both THCA A and B will decarboxylate to THC. Delta 9 THCA’s will decarb to delta 9 THC. Delta 8 THCA’s will decarb to delta 8 THC. Etc.
this might be more important if they ever start making prodrugs of thc. cause attaching functional groups without affecting activity might be easier on thca b
So, maybe an isomerization pathway??? This is getting a little too “in there” for me. Doesn’t surprise me that there’s variance. And variability found naturally. But the path to yield these would all fall under an isomerization of some kind.
Love the science here but what is the practical goal ?
Separation of these isomeric mirrors achieves what ?
Uses would have to include non combustion related process and delivery methods I’m thinking a transdermal or sublingual application would be the only product route for this achievement
THCA is good for the skin but sure is spicy when orally consumed
the one with the acid on the bottom decarbs at a much higher temperature because theres hydrogen bonding coming off the hydroxyl which causes the regular acid to decarb at a much lower temperature, i believe @mitokid told me this
in theory a selective decarb could allow you to decarb mostly 1 which would allow seperation by column chromatography
kinda how cbd and thc can be partially decarbed and seperated