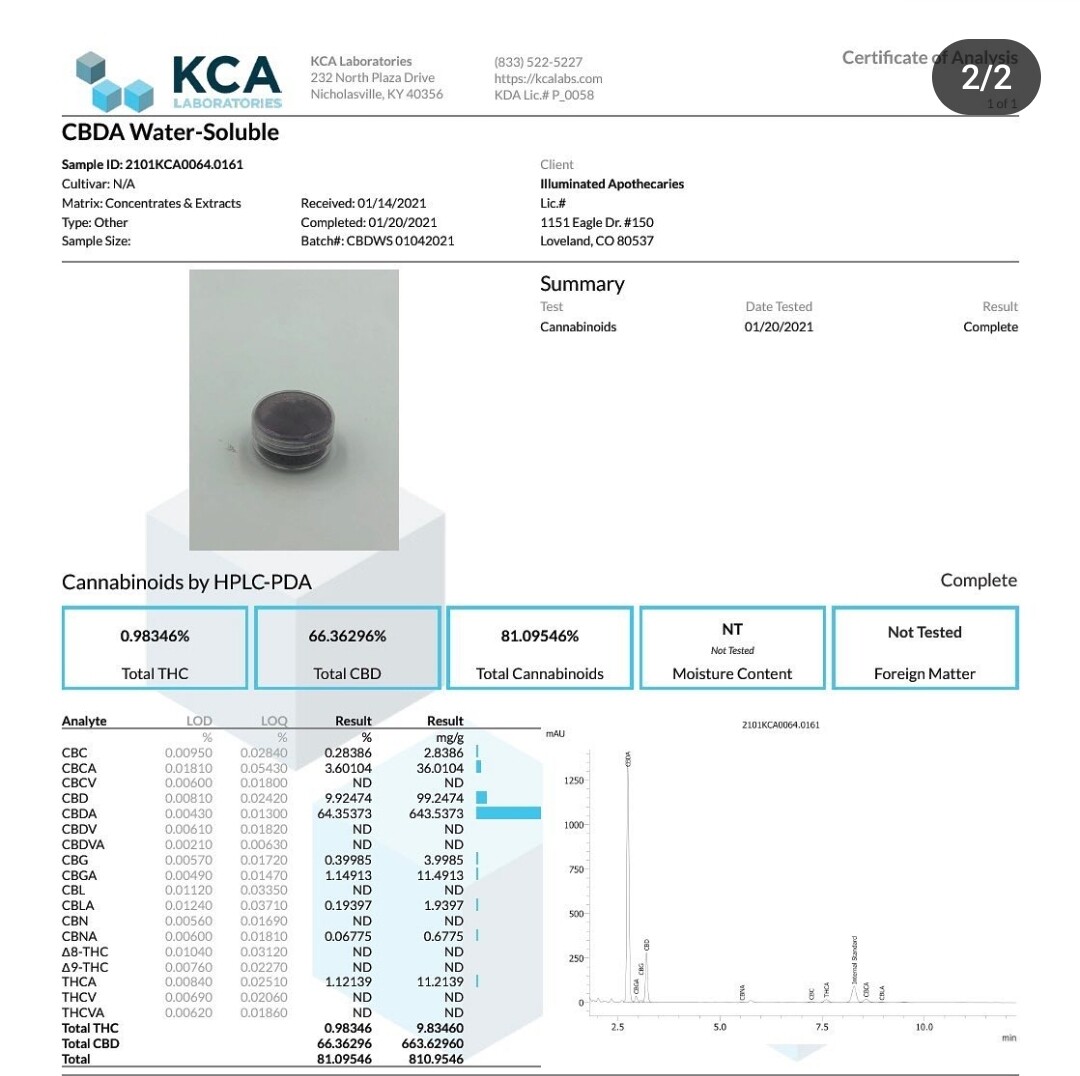
So I was doing some reading and brushing up on some fundamental chemistry. I have found that sometimes while reviewing topics that you know, things that you may have missed the first time around reveal themselves. This kinda feels like one of those moments.
Hydrocarbons are a great solvent of choice because like dissolves like. The target molecules are non-polar and the solvent being used is non-polar. So I started thinking, the target molecules that we are extracting are acidic. So could you in theory adjust the pH of the water and effectively extract cannabinoids?
I realize that adjusting pH is a common practice in water washing completed distillate for purification. But what if you protonated water prior to decarboxylation to extract the cannabinoids from the biomass? Would it actually extract the target cannabinoids? Has any one explored this? Would this still be considered a solventless process?
This is also a semi-synthetic method as you temporarily strip hydrogen from the -COOH bond and replace it with a metal - making it water soluble (semi-synthetic molecule), then later remove the metal and replace the hydrogen from a new source (an acid) - to return it to its “original-ish” state. Not much different than washing olive oil from your hands with soap - with one extra step (re-acidification).
Nothing is perfect. Residual acid / base may exist as well as side-chain reactions.
Sounds like I need to read a patent or two and then submit an R&D proposal. Not gonna lie, equally excited and disappointed this area has already been explored.
I don’t think any chemist would call simply deprotonating something “semi-synthesis” – if that’s the case I guess I’m “synthesizing” my margaritas when I add some lime. And then after that my stomach is doing some more synthesis! A little bit of a stretch if you’re trying to claim it’s somehow something bad.
Hell, since THCA is a “weak acid” (meaning it will deprotonate in neutral water to a small extent), you’d have to say even just mixing oil and water is “synthesis”.
You really have no idea if you are stripping a “hydrogen from the the COOH bond”…because as noted by myself on dozens of occasions “Butane works no theory” because the state of the Carboxylic acid in the trichome storage solution volume element (perhaps two separate phases of different polarity…etc for those who understand) is actually unknown.
Butane is unique discussion on it own, and it certainly works well. Extraction with ethanol which is protic , probably yields an equilibrium mixture of anionic form(s).
If you read past posts here, you will find I have noted that many anionic forms may exist…depending on solvent polarity , water content, pH considerations, and non aqueous solvent effects on pK values, intramolecular hydrogen bonds and intermolecular hydrogen bonds with solvent and water.
Let us say when increasing pH with base, after pH 6 the COO- is prevalent and solubility of THCA in aqueous increase. Recent paper by Australian group provides an outline for this observed phenomenon.(Redirecting)
Further titration to pH 12 removes the phenolic hydrogen.
So one can totally change the cannabinoic acids to double, negatively charged anionic forms. (Water soluble)
the strategy of extracting cannabinoic acids with base water may well transform the industry for all those uses of cannabinoids that do not require the “taste of terpenes”.
There is a considerable ignorance in general when it comes to precipitating Carboxylic acids as neutral forms, salts and complexes. You might have a nice white powder or crystal, it may test out at 99% for one reason or another , but what the exact nature of the original crystal you sent for testing may remain unknown. (DOI: 10.1021/mp060126o and DOI: 10.1021/acs.cgd.8b01159): just something to keep thinking about.
When you see a nice HPLC chromatogram of THCA for instance. You see a nice “single peak “ on the chromatogram….what do you “think” that “peak” represents in “molecular form?”.
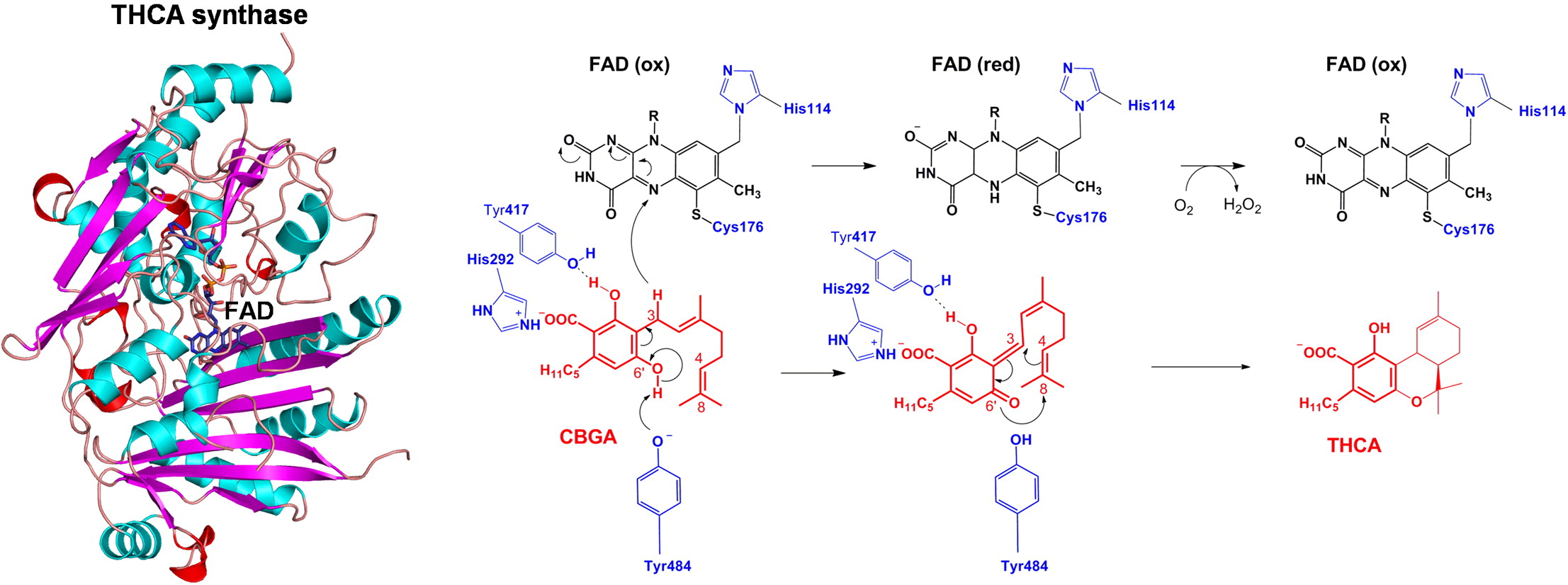
Which is back to @Zack_illuminated original statement about “stripping” protons. I am suggesting that in situ aqueous phase of the trichome storage volume the pH is such that the protons are already “stripped” and THCA-anion forms and or complexes may predominate. The latter seems to be the case when the recently synthesized molecule is released from the synthase. Zach I’m only pointing out that there is no obvious reason NOT to think the biochemistry is otherwise.
Adding HCl to a molecule then removing the HCl without actually changing the molecule would remain “organic” per-se. Just like salting psilocin with furmaric acid. The molecule is still psilocin and not isomerized in any way (changing of a molecules shape without adding / removing anything) - just adding HCl next to the molecule then removing it from next to the molecule. The molecule is still the molecule and never was anything else. To which I have seen plenty of people arguing simple CBD → D8/D9 is semi-synthetic just as an isomerization so idk why complete removal and re-addition would not be considered semi-synthetic.
But when you completely strip off hydrogen that was naturally sourced from the cannabis plant, then replace it with one from an acid, then you are no longer an organic molecule as far as I am concerned as it is no longer 100% naturally sourced from its host (the cannabis plant).
This process cannot be properly completed without the use of a hydrocarbon for the washes so its kinda moot either way.
As for which site is most likely to react would be the lone -COOH bond, which is very well known to react with bases to form salt and water such as washing olive or vegetable oil off of your hands with soap and water as they are both carboxylic acids. Reacting a more stable hydrogen is harder to do and would require elevated conditions - if possible at all and you might isomerize first.
IDK, I am no chemist, but when a molecule gets modified like this, I would completely see it as semi-synthetic as you are adding outside molecules to the molecule. To each their own I suppose.
This process has a ton of waste, consumption, and is messy. Much easier (and cheaper) methods already exist.
Have got a ton of experience with this process from when I was making CBDa water soluble (zero emulsifiers) a few years back but gave up on it because it was semi-synthetic. Basically an acidic alternate to HU-331 that is a water soluble salt.
True, but it’s semisynthetic in the same way decarboxylation is semisynthetic. Honestly not sure if that term is a useful descriptor or not for the process you are describing… I guess I would lean towards agreeing with you since the analogous chlorophyll copper complex is i think usually described as semisynthetic
Not quite the same… loss of CO2 is something that was already there.
Nothing is being put in its place - even temporarily during the process - then consequently removed and replaced with a non-plant derived molecule.
Just like putting THCa in water, yeah it might deprotonate but until you react it and actually replace the hydrogen, no reaction has taken place. As soon as you remove the water, the THCa will re-protonate naturally and nothing has changed.
That is no different than salt in water… acts the same way…
I’m not sure this is what defines whether something is semisynthetic or not. I’m splitting hairs here now, but wouldn’t you consider decarboxylation of serine to ethanolamine (or any other amino acid) to be semisynthetic? Not that this website needs more useless nomenclature discussions lol
When you take a naturally sourced plant molecule such as THCa/CBDa (C22H30O4) and remove the hydrogen from the -COOH bond by a base such as NaOH (drain cleaner) you get C22H29O4Na (*semi-synthetic molecule), then consequently react it further with an acid such as hydrochloric acid {another drain cleaner} you get (C22H29O4Na + HCl = C22H30O4 + NaCl) and create another semi-synthetic molecule along with table salt as the byproduct…
but of course there will be water in there also because you are not using pure HCl .
I need to go back and re read that thread. I think that a molecule inside of a synthase enzyme is not necessarily going to follow pH rules while inside of the enzyme.
After synthesis if the molecule is not soluble in water why would it deprotonate? Am I remembering wrong that the THCa enzyme specifically makes a protonated form?
It makes sense to me that the THCa is very very very slowly deprotonating in the basic environment because there’s relatively small amounts of basic water even available to dissolve into AND THCa isn’t readily soluble in water in the first place, which it needs to be in order to be efficiently deprotonated by the basic conditions.
depends on the activation energy, ΔE ‡. According to the Boltzmann distribution the proportion of water molecules that have sufficient energy, due to thermal population, is given by
where k is the Boltzmann constant. Thus some dissociation can occur because sufficient thermal energy is available. The following sequence of events has been proposed on the basis of electric fieldfluctuations in liquid water.[10] Random fluctuations in molecular motions occasionally (about once every 10 hours per water molecule[11]) produce an electric field strong enough to break an oxygen–hydrogen bond, resulting in a hydroxide (OH−) and hydronium ion (H3O+); the hydrogen nucleus of the hydronium ion travels along water molecules by the Grotthuss mechanism and a change in the hydrogen bondnetwork in the solvent isolates the two ions, which are stabilized by solvation. Within 1 picosecond, however, a second reorganization of the hydrogen bond network allows rapid proton transfer down the electric potential difference and subsequent recombination of the ions. This timescale is consistent with the time it takes for hydrogen bonds to reorientate themselves in water.[12][13][14]
The inverse recombination reaction
H3O+ + OH− → 2 H2O
is among the fastest chemical reactions known, with a reaction rate constant of 1.3×1011 M−1 s−1 at room temperature. Such a rapid rate is characteristic of a diffusion-controlled reaction, in which the rate is limited by the speed of molecular diffusion.[15]
I’m sorry but you’re just wrong. No chemistry textbook in the world would ever support a claim “deprotonating and then reprotonating a chemical makes it not organic”. As far as you saying they “replaced” H+ with Na+, that is actually not chemically accurate. COO- just floats around in water clusters with occasional Na+ also floating around, no different from the natural state of COOH in water, which is that there will be COO- surrounded by water clusters with occasional H+ bonded to the clusters. Calling it COONa is just a formality describing the balance of the solution as a whole, not a try description of what the molecules actually are in that solution.
I would ask you to ask anyone who works in chemistry, in any field in the world, whether they would consider an acid/base change, in water, less organic or pure or natural than introducing a non-water solvent to do it instead. I don’t think you can find a single person who agrees with you in 100 years. Maybe you can get Chat GPT to agree with you. I’m going to trust my MS on this one.
Boy, if you considered every reaction of an acid with a base as “non organic”, you couldn’t live a day doing any of the activities you do. I wonder if you’ve ever applied this argument to anything else in the world.
You need to go back and read the X-ray diffraction study with Look closely at the diagrams for cannabinoic acid binding to the enzyme. The X-ray data show the form in the cleft to be anionic. this is the 2012 paper and the figure published in this later study is different from the 2005 diagrammatic figure published in 2005.
You understand that the maximum rate for the synthase is achieved at about pH 5-7. We are talking aqueous protic-biochemistry here…THCA is quite soluble as anion at this pH. We’ve talked (cyclopath) the published “tea” data.
Trying to understand how butane works is an entirely different subject…because like you suggest it is difficult to get the neutral form into aqueous .
A two phase storage compartment may do the trick 1. first step where Intramolecular h-bonded anionic forms may partition from the aqueous phase into the “terp soup” phase…2. this transition may well effect a solvent induced pKa shift of the carboxylic acid moiety, such a shift may well rob the aqueous phase buffer of Hydronium ions at the interface. I.e., an interface arrangement of cannabinoics with their anionic moiety facing aqueous. Interface protonated neutral forms would then partition fully into terpene abundant phase. Thus a terpene solution phase of neutral THCA (H) would be in equilibrium with an aqeous protic phase of carboxylic acid anion (-) form. The cannabinoic acid in each phase having a distinct pKa while the aqueous phase maintains a single buffered pH value.
So I am not opposed to the concept of Butane extracting the “terp soup” phase at minus 40. It does not rule out that butane may also extract molecular complexes .
The microscopy of the trichome storage volume…shows globular regions which are discontinuous with something akin to an aqueous phase…
A two step process is ok with me.
You have to come up with a storage “form” that exits as a extractable liquid at -40 and is readily extractable in non polar aprotic solvent.
Think about he question I asked above:
When you see a nice HPLC chromatogram of THCA for instance. You see a nice “single peak “ on the chromatogram….what do you “think” that “peak” represents in “molecular form?”.
Everything you need to know is encrypted in the studies of reverse phase HPLC chromatography of cannabinoids.
What is wrong with that chromatogram? Someone who studied with Snyder, Kirkland or Karger will immediately tell you!