This technology is old now and always seemed obvious to me seeing how its the same schema as fractional distillation. and I’ve been working in co2 extraction for 8 years and keeping my ear to the ground for this sort of thing but still haven’t seen any good data sets on this topic. by good data sets, I mean one that shows how the pressure and temp settings of collector 1, 2, and 3 affect the compositions of the others downline
for alil backround
I remember that the extraction vessel was at around 2500-2800psi 122f for maximum extraction without chlorophyll which starts to dissolve at 2800-3000 +psi
so first collection-A pressure was right below critical pressure 1000 psi and at or near 95f
collection B was between 600-800 psi and 80f
collection C was 375 - 425 psi
most of the time i would we would just split two fractions the terpene fraction and the cannabinoid fraction or just the terpene+cannabinoid fraction and the wax fraction
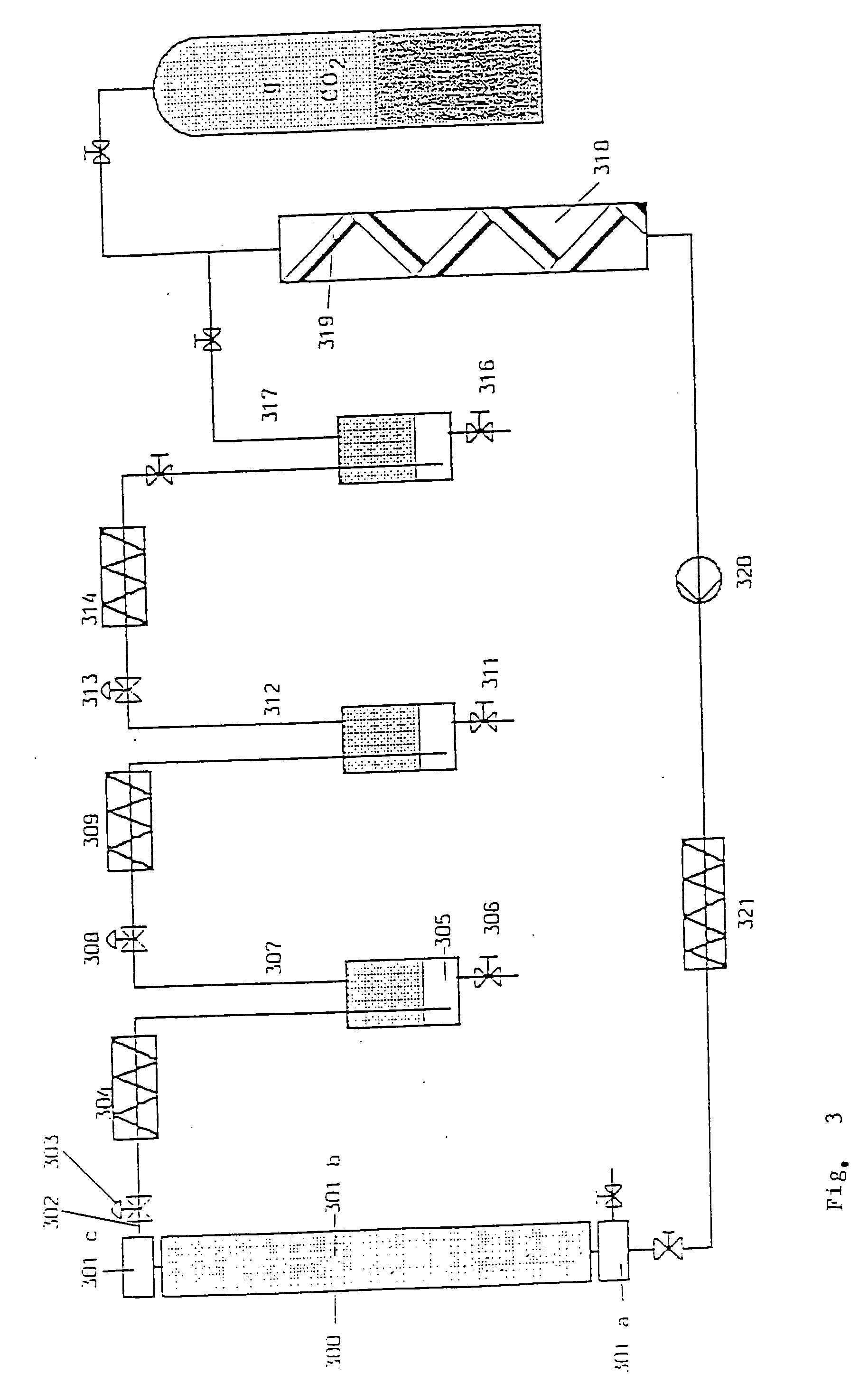
To this end, preferably a high-pressure column (FIG. 3) subdivided into segments, comprising a bottom segment for dissolving the primary extract in supercritical CO2, a purification segment filled, e.g., with silica gel (mean particle size of 0.02 mm to 0.2 mm, preferably 0.1 mm), a head segment for discharging the mixture dissolved in supercritical CO2 of CBD, Δ8-THC and Δ9-THC into three separating vessels for separate separation of the purified CBD and the purified Δ8-THC and Δ9-THC.
[0071]
The extraction conditions prevailing for purification in the column are supercritical for CO2, preferably 180 bar and 55° C., in the first separating vessel where CBD is separated out for CO2 subcricital conditions in terms of pressure and supercritical conditions in terms of temperature, preferably 70 bar and 50° C. In the second and third separating vessels, where Δ8-THC and Δ9-THC are separated out, conditions subcricital for CO2 in terms of pressure and temperature are to prevail, in the second separating vessel preferably 60 bar and 30° C., in the third separating vessel preferably 55 bar and 25° C.
There are several people working on this and some of the operational results are insane. Everyone I know that is doing the work are bound like shibari from sharing details…but it’s a worthwhile endeavor for sure.
Its funny cause i kept having the feeling like this was happening when talking to people but thought i was probably just paranoid. i know most companies dont share specifics but usually they’ll say something
I have run a waters sfe for about 4 yrs now and we have experimented with a couple different methods (pressure/temp/time) as well as some inline filtration tests. I am kind of embarrassed to admit that we have really only ever run supercritical phase extractions but have begun to recently play around with lower pressures. I’m happy to share some of our methods and results. What type of silica and would the use of the silica be based on a percentage of silica to raw materials?
@Plant2pipe. We tried activate carbon both on top of the raw materials and also is a separate column (v1 to v2). We had some limited success however I started researching “cake” filtration saw that due to the pressures that we run 3,000 to 5000 psi the media would channel and not really perform as anticipated. I like the idea of trying Silica gel 60.
Our work around was to build a separate pressure filtration system whereas we run our winterized solution (4x 1 ethanol to extract) housed in a pressure vessel with a dip tube and back filled with nitrogen pressure. We make a slurry of charcoal and eth and the use a filter stack (10 micron and 2 micron filter) and fill a 18 in column. Solution is run through column and comes out pee gold the n the other end. Charcoal use is anywhere from 8 to 10% of starting solution volume. Psi ramps up from 20 to 50 or 60. 3000 ml is about 4 hours.
I am definitely going to try the silica gel 60 and report back our results.
Ps. Don’t mind the mess. A couple charcoal blowups in the beginning.
love the idea and the implementation, have you tried using co2 pressure? ive read co2 will reduce viscosity which makes filtration easier. idk if this will do anything unless at a higher pressure so there is some liquid co2 in contact with the extract but might be worth a try
Thank you kind sir!! I was so tired of Büchner funnels that’s for sure! I need a more efficient filtration set up as this just isn’t keeping up with our current needs. Any suggestions? What system do you operate on?
I was intrigued by the idea of running the solution back through the waters machine although I’m pretty sure I would need to change the flow direction on the system. I.e. co2 Flow down. Also I’m not sure about leaving one of the collection vessels open to make it a dead end/open end operation. I guess is would work though. Hummmmm. Now you got me thinking.
Can you post a picture, or maybe you already did, of your crude non-dewaxed and dewaxed in EtoH? It’s hard to gauge the quality of the filtration without a before and after.
@shinyemulsion I will post some pics tomorrow of extract. Straight out of the waters SFE and then winterized solution and after final filtration. I have a pic of the filtered solution up but will show a before pic also.
You can separate CBD from THC on a Waters SFE but the 3rd cyclone can’t go below pressure of the rez that sat at 750psi at least:/ I’ve never got any good answers from some of the bigger people on IG for my custom endeavors; usually when the conversation would end😬 I’ve always wanted to see about it with more cosolvent fractionation. Propane I thought would be cool cosolvent, but never knew about safety until I saw Jonarronbrays sweet post🙏