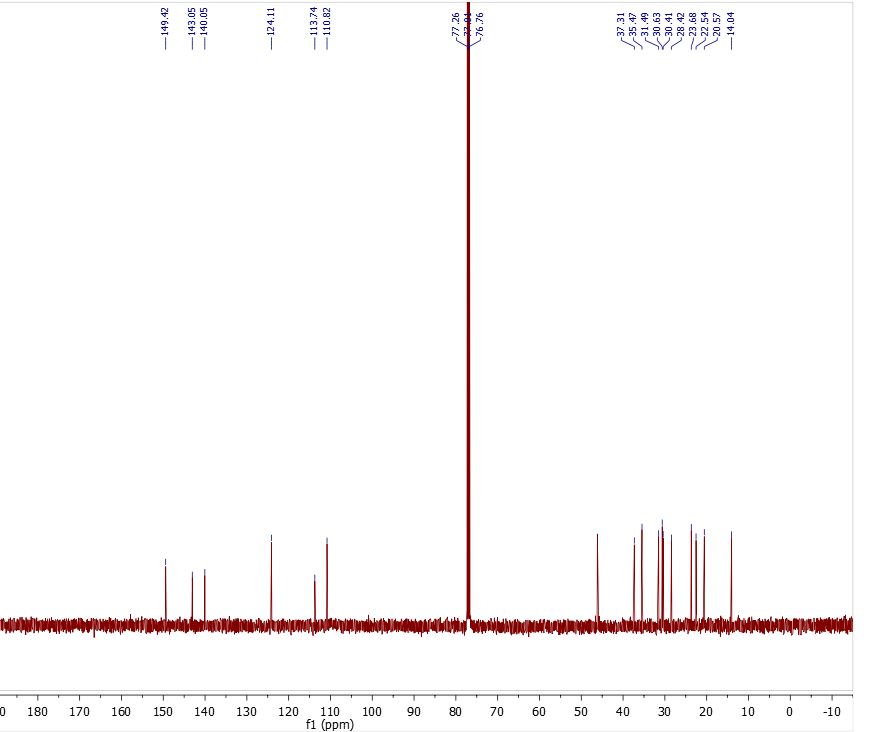
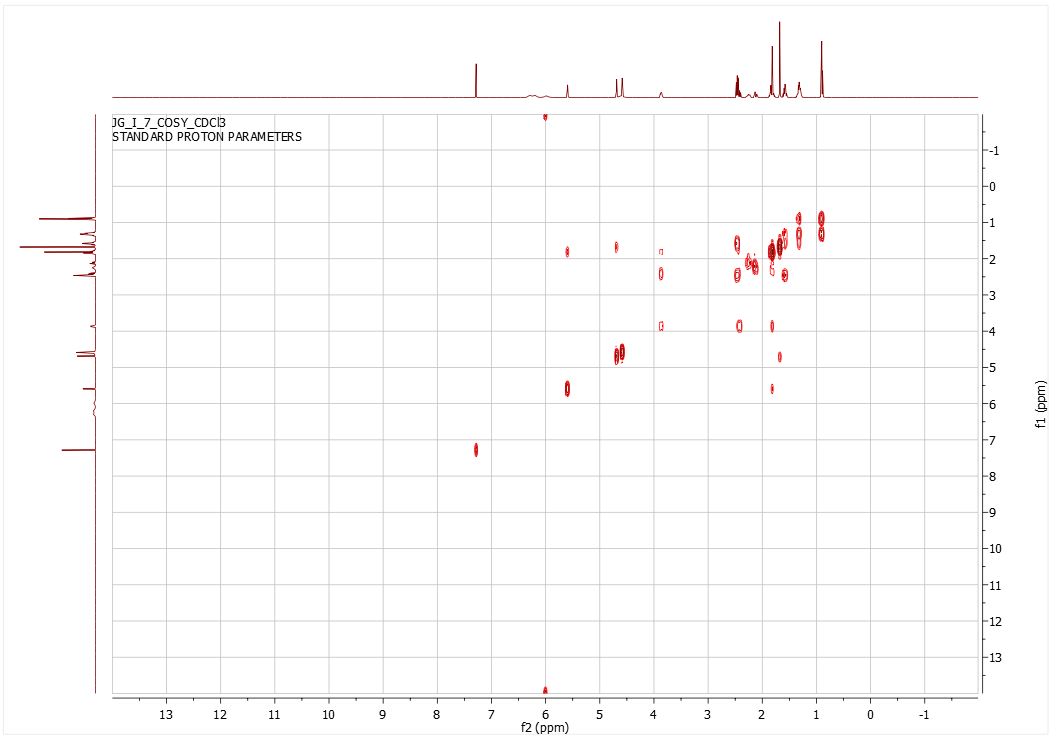
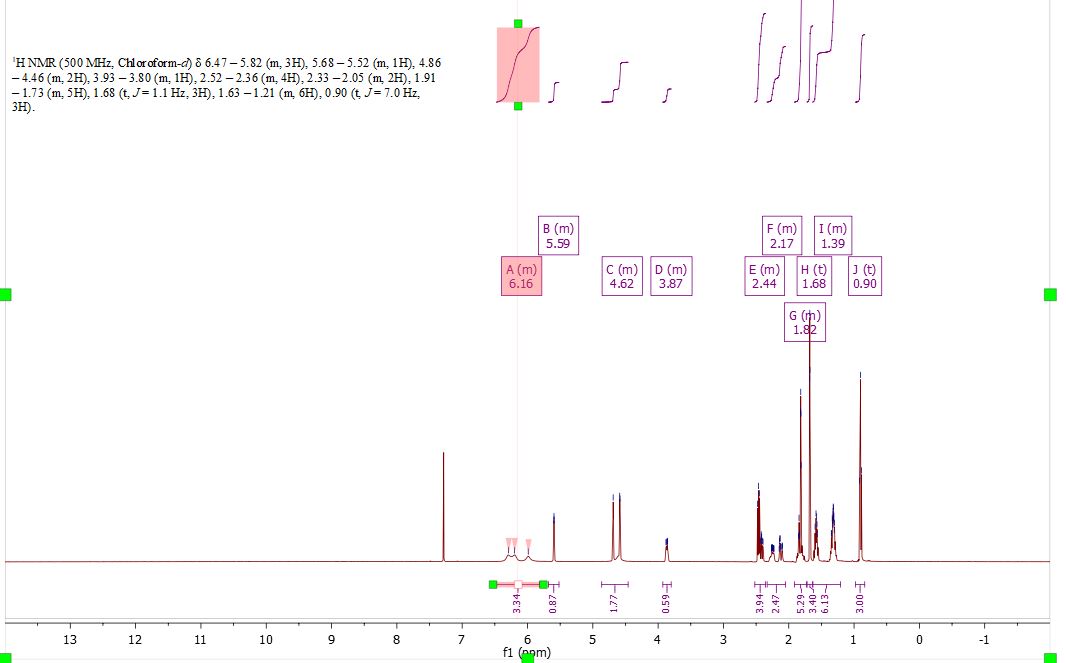
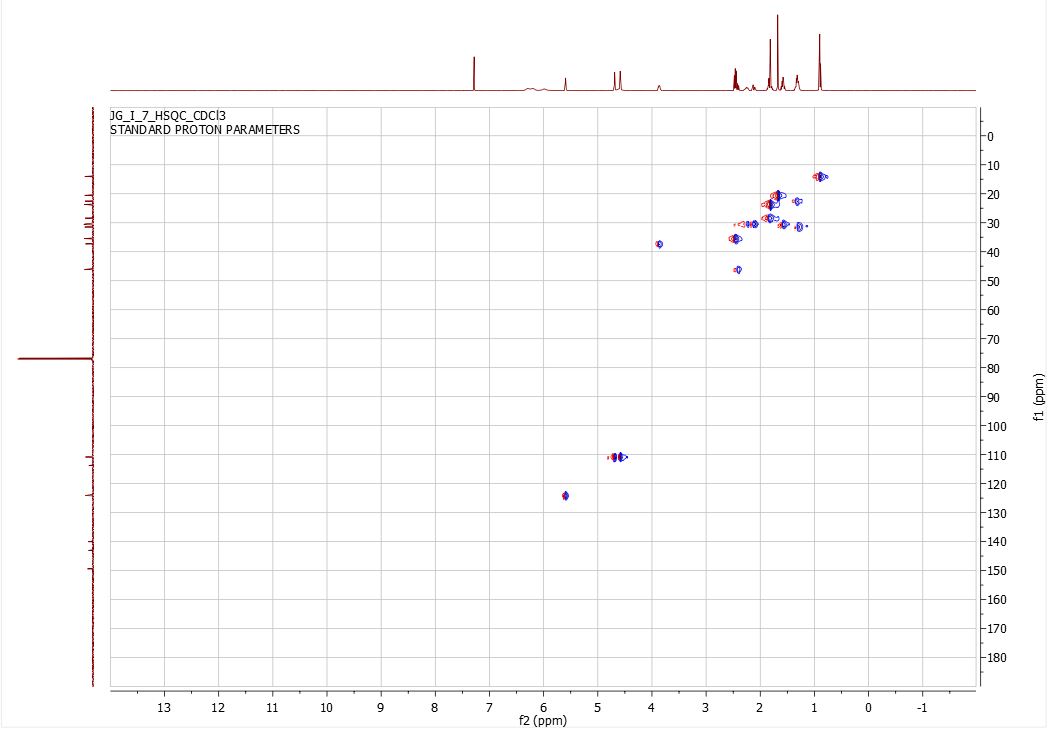
Here is some recent data I compiled on proving the structure and purity of the CBD I have produced on industrial scales past…
Name another chemist with HSQC and COSY data of their isolate lol
From the top down,
- 13C NMR
- COSY
3.1H NMR
4.HSQC
Here is some recent data I compiled on proving the structure and purity of the CBD I have produced on industrial scales past…
Name another chemist with HSQC and COSY data of their isolate lol
From the top down,
Haven t gotten a cleu how to read these but i guess that huge Spike is ggooood
But hè be proud ![]()
![]()
lol the spike is prolly solvent
Told You ![]()
![]()
![]()
![]() don t understand
don t understand ![]()
![]()
What kind of purification did you use to get your product this pure? I also think you should label this spectra for others to understand that all of the protons and carbons are present in the spectra.
This is awesome thank you for sharing
If you desire to learn nmr spectroscopy you must be self motivated, I compiled the data/spectra I wont spoon feed the rest to you damn ingrates lol
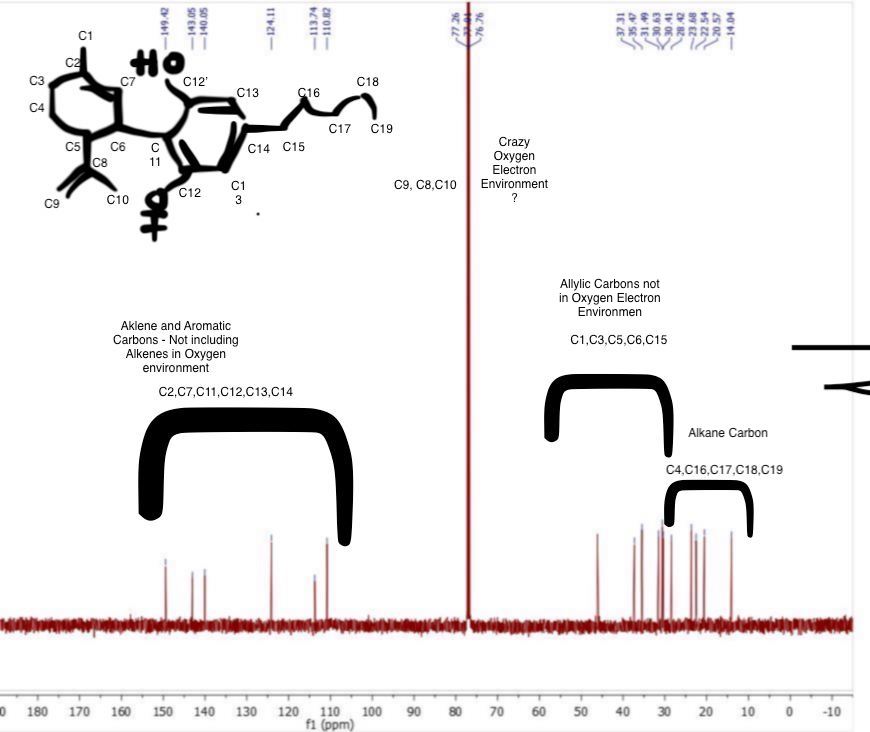
First guess, clearly my undergrad education won’t get us much resolution and I don’t have a great explanation for the 3 carbons so close to one another around 80.
We could probably google this but where’s the fun in that.
It is the CDCl3 peak. The 3 signals are the carbon being split.
https://webspectra.chem.ucla.edu/NotesOnSolvents.html
@QGA The multiplet A at 6.16 integrates to 3? So there are 3 hydroxyl groups? Is that a spectrum of CBDA or some temperature dependence?
For THC NMR this was a good read:
Thankyou! I was pretty sure that’s the solvent but I can’t get the Carbon # to match up. I count only 16 peaks in the CNMR and 19 unique carbons as labeled. What am I missing?
If I remember correctly there are three lines because deuterium has three spin states.
Well C13 and C1 on the aromatic ring as you have it labeled are virtually chemically equivalent. The signals in the alkyl chain are probably really similar too. There isn’t really a good way to get integration from C13 spectra so it is not possible to tell how many carbons are coming into resonance at that frequency.
Makes sense! I had no idea carbons could be on top of each other like that.
C1 on the Aromatic ring is a typo and is meant to say C13!
Yes this peak was interesting to me, either the program is a bit off for integration (mestrenova is the software) which is common but also they may be trace of cbd-a or some other small contaminant. Could be a result of the two hydroxyl groups ability to have rotation about their axis via the shared carbon ortho to them both. Putting them in more than two chemical environments during the NMR time scale.
Did you use that last peak as the standard for integration? I don’t know what the actual name for it is in the software. Just asking because that one integrates so nicely. But I guess that is the peak for the end of the pentyl chain so it is a definite 3.
For sure there is a chance it is sterically hindered rotations. Its pretty bulky. You could just take the spectrum at a higher temp to prove whether that was true.
Anyways this is really cool so thanks for sharing.
Yea might do it, but not horribly focused on it right now, it fits and ships for my purposes I was mainly curious I am not publishing just helping someone at my university publish. If I am co-author i will make sure to share!
carboxylic acid protons are typically found from 12 to 14 ppm. The three protons at 6.16 ppm are the protons from the olefin.
Yeah carboxylic didn’t seem correct. And so I just went and looked and you were right. It was on the damn paper I linked. It really looked like O or N because of how the peak is broadened. And then you look at C and D and they are OH with sharp peaks.
Does (m) stand for multiplet in this case? I also see (t) which normally means triplet, but the labeling seems off.
@QGA You got coupling constants? I know you just said it wasn’t at your discretion to share though. ![]()
S is singlet, d= doublet, t= triplet, m=multiplet, dd=doublet of doublets, etc. From my experience, I have rarely seen NH or OH protons in chloroform due to rapid exchange. You can best see OH peaks at about 5 ppm when using DMSO as the deuterated solvent for NMR.