I’ve been doing about 15 minutes but room temp. My sonicator does not have temp adjustments.
1 Like
@Cassin I know some chemists on the board. However I am not a member so this must have slipped my radar! Thank you, I see the membership is $60 and OMA is 120… so for 180 I can get the full prep and papers discussing the reasoning? Worth it if that is truly what is available.
100% yes.
And all the drafts.
And come to the meetings and meet the others doing this work.
The AOAC member fee gives you access to the entire compendia, not just the cannabis stuff. I use it for all my other product testing standards and things. Since my products don’t just have cannabis in them.
And the ASTM fee gives you access to all the D37 committee work AND one other set of work you are interested in. I picked tubing and pipe standards - since I use those all the time for equipment design/fabrication activities. But there are thousands of others you can pick from.
But this isn’t about becoming a member (its awesome if more people do, the more diverse voices the better) - this is about answering his question.
Here’s the prep method digested into a bullet list…
For flower and kief products:
- Start with 5grams and homogenize down to particles of less than 1mm (cryogenic homogenization is recommended, using liquid nitrogen then homogenizing the particles down to less than 1mm) store these frozen before extraction
For liquid products (oils, tinctures, etc):
- Vortex then store refrigerated before extraction
For flower samples:
- 0.50g of sample into 50mL tube + 20mL EtOH
- Vortex the sample briefly
- Shake sample using a horizontal shaker for 30 minutes at 250rpm
- Centrifuge at >3000 g for 5 minutes then filter (0.22um) and keep back the sample.
- Repeat these steps twice - both 20mL supernatants are collected. in a 50mL volumetric flask
- Dilute sample to volume in the 50mL flask
- Pull an aliquot and filter this - dilute with this aliquot, leaving the beginning sample for any future dilutions.
For all other non-flower samples (but not edibles, that’s a different process especially chocolate! all edible matrices should be validated separately to verify no interference from any non-cannabis stuff):
- Weigh 0.5g into 25mL flask add 20mL EtOH
- Sonicate sample until samples is fully dissolved. (In practice this doesn’t take very long…like a minute or less)
- Pull your aliquot and filter (0.22um). Use this aliquot to make your dilutions. Save the rest for future tests additional dilutions. Keep in the fridge.
2 Likes
Thank you for breaking it down as you did, very good info! This caught my eye
This seems like an awfully high starting sample amount, is it not? I have heard of some labs requesting 1/8s of flower for testing but that always seemed excessive to me.
I also wonder what the purpose behind the centrifuge step?
Mentions briefly here, can we add in an appropriate time frame? I’ve been vortexing for 90 seconds absent any other information
2 Likes
This helps! Thoughts on MeOH vs EtOH?
For homogenization freezing this will keep the kief from sticking to weigh boat/ mortor pestle (assuming this is breaking down particle size efficiently?
So if I understand correctly…
- 0.50g of sample into 50mL tube + 20mL EtOH
First stock is 500mg/20ml - 25 mg/ml or 25,000 ppm. Which is a 25 fold dilution. - Vortex the sample briefly
- Shake sample using a horizontal shaker for 30 minutes at 250rpm
- Centrifuge at >3000 g for 5 minutes then filter (0.22um) and keep back the sample.
filter the entire 20 ml? or smaller aliquot? - Repeat these steps twice - both 20mL supernatants are collected. in a 50mL volumetric flask
- Dilute sample to volume in the 50mL flask
by both you mean …?
20ml into 50ml…0.4 dilution? - Pull an aliquot and filter this - dilute with this aliquot, leaving the beginning sample for any future dilutions
**Dilute to achieve ppm on curve?
final dilution 25.4? Clearly my logic needs help. I appreciate all the effort!
When you make your dilution you have a final ppm in mind relating to the curve? ~2000 DF is what I have seen before
Maybe something like this may be helpful to you?
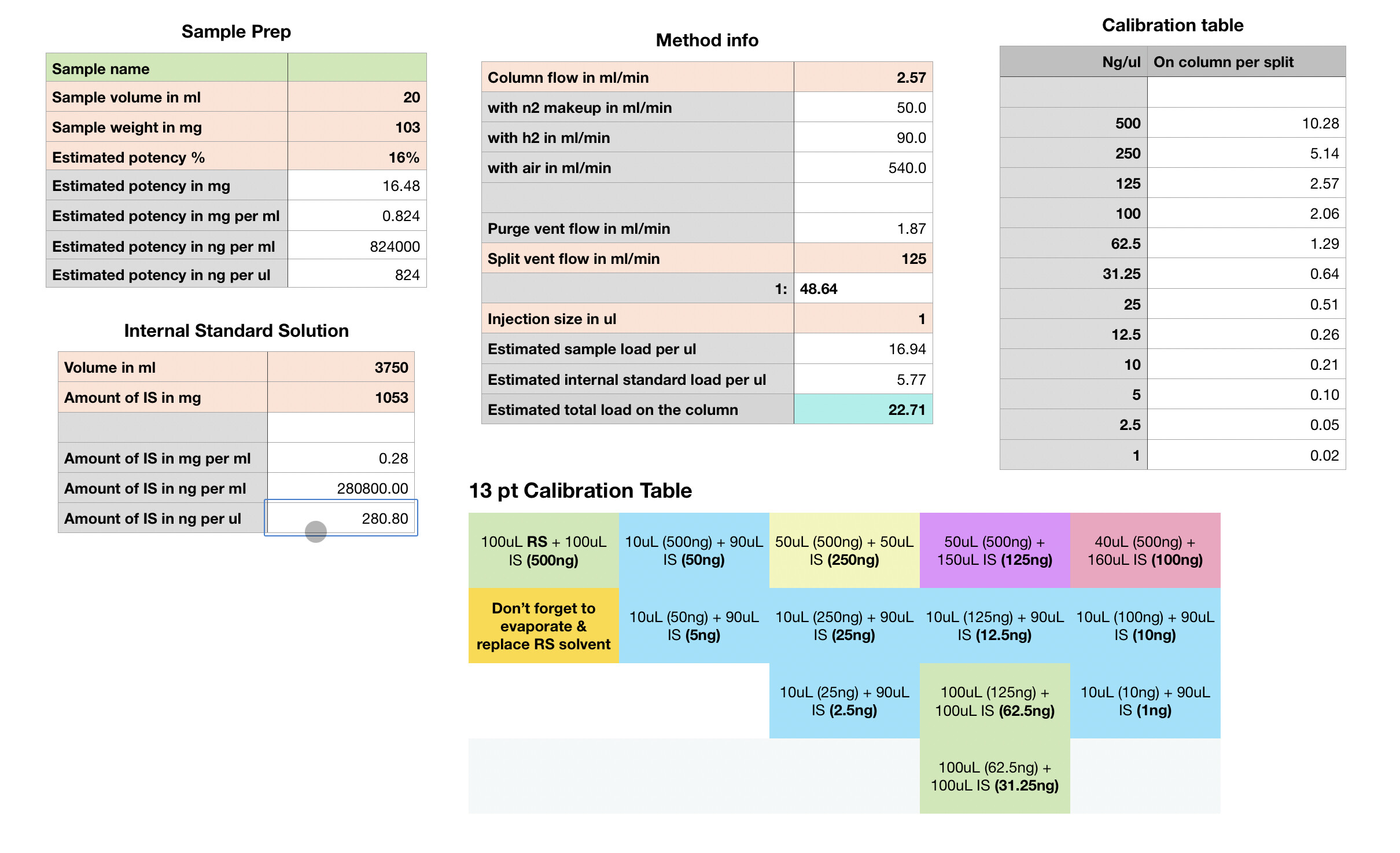
With this spreadsheet I made I can play with the numbers regarding sample volumes, weights and get an idea of what kind of load on column should be expected based on those settings. This also helps me understand if which calibration points are closest to the estimated load.
I’m pretty new at this so please take it with a grain of salt…
5 Likes
There’s a pretty good paper on this that is referenced in the AOAC guide. I don’t have a copy to share with me unfortunately.
The moral of the story on this was specific to recovery percentages and sample stability.
Ethanol pulled out more so there was a better recovery percentage. Methanol was close - but had less sample stability, which is mostly due to methylation of the active compounds. Methanol is also a harder chemical to handle for waste disposal and for operator safety.
In a volumetric flask - you add whatever your a mixing together and then you fill the rest of the volumetric flask to the line. So you’ll end up with exactly a 50mL solution, which has the 20mL + extracted substances x2 plus whatever additional solution you needed to get a total solution to 50mL.
When you do this you don’t have to wonder about the stock dilution numbers. You establish that you have X cannabinoids in 50mL - and you’re using the instrument to tell you want X is - and then you say, there was X cannabinoids in 0.5grams. This is for prepping samples not standards. Does that make sense?
And yes - dilute to achieve expected PPM on curve. And to make sure that your peak is never more than 150% of your upper calibration point.
There are a lot of ways. That way would be super slow. I’m going with bb’s in the tube after its frozen and vortexing it for 45 seconds. This has worked for me. I know there is commercial tek that does this work now as well. and I’ve also seen people have good success using different kinds of grinders - although I think those are very hard to clean. BB’s are cheap enough to throw away - you know?
I target my dilutions to get me at around 100ppm. That’s the mid-point on my calibration scale and so its a sweet spot. If the sample runs and is more than my upper point, then I dilute again. I start with normal sample, then 10x and 100x dilutions. That works for most things - not heavy concentrates and not isolate. Those need more dilution. -shrug-
2 Likes
I am in a similar boat so I won’t judge. This is in way more detail but I do something similar (the goal anyways). Always helpful to see what others are doing! Are you using Hydrogen as your carrier gas? Separation seems to be good and I got it down to a 6 minute run. I’m only looking for THC CBD CBN and CBG. I have a 4 point calibration. 13 is impressive. Have you found the IS help a lot/necessary?
So for the prep your doing 100mg/20 ml at 5 mg/ml - 5000 ppm. How are you calculating expected? Is this from flower that you are testing against or is this a theoretical vs actual situation?
1 Like
Don’t mind the numbers on that spreadsheet too much (as entered anyway), mostly was playing with figures.
I run a RXI-35sil MS using the following settings (as taken from Restek):
2.5ml/min h2 column flow
125ml/min split vent flow
N2 makeup at detector to 50ml/min, h2 to 90ml/min, air to 540ml/min as measured by volumetric flow meter (my equipment is old and has none of the bells and whistles)
I can’t really comment on the rest of your questions at the moment because I had a seal blow contaminating my gas lines ![]()
I would say the calibration table as-is is excessive and unnecessary for the most part
Got it. FYI if you aren’t using the mass spec heard a secret that it is unnecessary feature on the column for FID and may be an upcharge.
1 Like
Funny you say that, I do have a mass spec model in my garage waiting to be setup and it does have all the fancy EPC stuff
1 Like
That makes sense about bb’s. So you are freezing and homogenizing the whole 5 grams you suggested and then sampling from this after vortexed with bbs? I actually incorporate 4 small stainless bb’s in my flower prep but when I have already sampled. tried grinding but did a test with just picking off pieces of the buds from different areas. Seemed to get a better potency this route rather than grinding. I think because too much loss of trichomes in the coffee grinder I was trying.
Crazy how scientific vendors try to sell you ceramic or stainless homogenizers for hundreds of dollars when 1000 bb’s on amazon is like $20!
Meoh or etoh are both really good solvents for cannabinoids. But etoh is a controlled substance and comes with a pretty hefty tax. Also Meoh is one of our mobile phases for lc so it was an easy choice. Also I find Meoh solutions tend to filter and pipette better.
ACN helps with flower extraction somehow, maybe more penitration because of change in surface tension or some other solvent properties. But the acn/Meoh mix does a bit better on flower extraction than just Meoh or etoh alone. This is not really needed for extracts because you are really just dissolving them and not extracting, Meoh is fine for them.
And ppm target is about the mid range of your curve, so probably 100ppm. So that is 20% flower diluted 1/2000 gets you to your midrange. Pretty good general dilution scheme for flower.
2 Likes
I’d recommend either making your own method from suggestion and trail and error, or paying for a consult. Trying for some n2 cryo grinding method designed by astm armchair is going to be a wild goose chase like no other and waste a bunch of your resources. My opinion though… Do what you do
1 Like
Methanol appears to be the most efficient extraction solvent for pulling THC-A. ACN appears to be 3rd according to this graph I found in an article that I’ll provide at then end of my post

Medical Marijuana Solvent Extraction Efficiency – Potency Determinations with GC-FID (restek.com)
1 Like
Meoh does not perform as well as acn/Meoh in my side by side extraction on +20% flower (6x replicate)… This is for acid forms. The difference was minor but notable. Correct that pure acn is not the best. Correct that pure Meoh is the best… Together they have synergy and seem to be best at flower extraction. There is a paper going into this synergy but I had to prove it to myself.
No I don’t have the data to back this up… But you can try it yourself if you want proof. Just make sure you homogenized your samples so you are not measuring variances across buds. And does that graph say 4% dry wt thca? That is not a real world scenario if so.
2 Likes
Here’s the sample sizes they were using: “0.2g ground samples (except in the case of acetonitrile where we ran out of marijuana and only used just over 0.1g) were extracted with 40mL solvent”
I can see both solvents combined performing a better job, so I’m not being a detractor. I just linked an article from restek, not trying to debate your real world experience. I was just adding more information to the post👍
3 Likes
5g is excessive and good luck fitting it all in a 50ml tube with enough room to have the ball mill do anything. You can get a very representative sample in 0.5g by just breaking of 0.1g from 5 different buds.
Sonication at elevated temp is important imo to get all variables tipped in the right direction. Which you want all the help you can get when extracting high potency flower at 1/10 extraction.
Sonicate 5min vortex 10s, repeat. Centerfuge is unnecessary because the particulates settle fast and you are filtering anyway… Only need to filter like 2ml btw
2 Likes
For clarity: Adding 0.5 ml to 5 ml is not a 1/10 dilution. This is a 1/11 dilution because the final volume is 5.5 ml.
For a 1/10 dilution, start with 0.5 ml and bring it up to a total volume of 5 ml with the diluent… now your final volume is 5 ml, in which your initial volume of 0.5 ml has been diluted 10-fold.
Same answer if you are starting with flower tissue: bring it up to 10 volumes of extraction solvent, instead of adding 10 volumes of the extraction solvent.