Much thanks in advance for all providing help and thank you to everyone who contributes often on future; you are all invaluable resources.
We have been dealing with coming and going issues of isomerization in our distillation process. I have dug through threads for months trying to see where others issues are similar/different, as it certainly is a well discussed topic. End product goal is high potency THC distillate. Here is our standard procedure:
Grind material
Decarb @ 240 F for 1:30
Supercritical CO2 extraction–> approx 1800 psi, sep temp 35
Dissolve crude into 10/1 ethanol; sit overnight @ room temperature
First coarse filtration @ room temperature; into -80 C freezer for 24 hours; perform 2nd coarse filtration
2 AC polishes filtered over beds of silica (top) and T5 and 1.0 micron filter paper
Additional filter of .45 micro filter paper
Rotovap to remove solvent
Perform Short Path Distillation using 2L Lab Society Kit . Gl-18 connections, typical vacuum depth approx 200 microns 1st pass, 120 microns 2nd pass. Chiller 50 C, vapor temp goal of 157. Lowest we’ve been able to get vac to is 75 microns. Typical 500 grams batch is in mains portion for 5-6 hours. Temps/vac are watched closely and rarely fluctuate.
*Distillate stored dark @ room temp with argon backfill
LLE using hydrocarbons is not an option for us due to our state’s regs unfortunately.
We began seeing isomerization pop up on COAs approximately 6 months ago. Initially believing media was making its way to the boiling flask and causing reactions, we improved our filtration process and upgraded equipment. We saw big improvements and the problem essentially disappear on COAs. Within the last month or so the problem has resurfaced despite these changes.
Questions:
How big of a role could ph play in causing these reactions? We are seeing 3-4 ph reading on post processed oil/ethanol mixture pre rotovap. We have attempted to raise the ph using magnesium oxide/water and have seen the ph get to neutral, but end up with what looks like a cloudy mess. Distillation of said mess ended with a messy boiling flask and reaction in general, and distillate with specs of debris, color looks good otherwise.
Tips for adjusting PH/confirmation about our PH adjustment process if it is a potential culprit.
Seems like several CO2 processors have dealt with these same issues with distillation; anyone operators have any consistent, repeatable solutions?
Any other ideas about potential culprits/where we can eliminate variables.
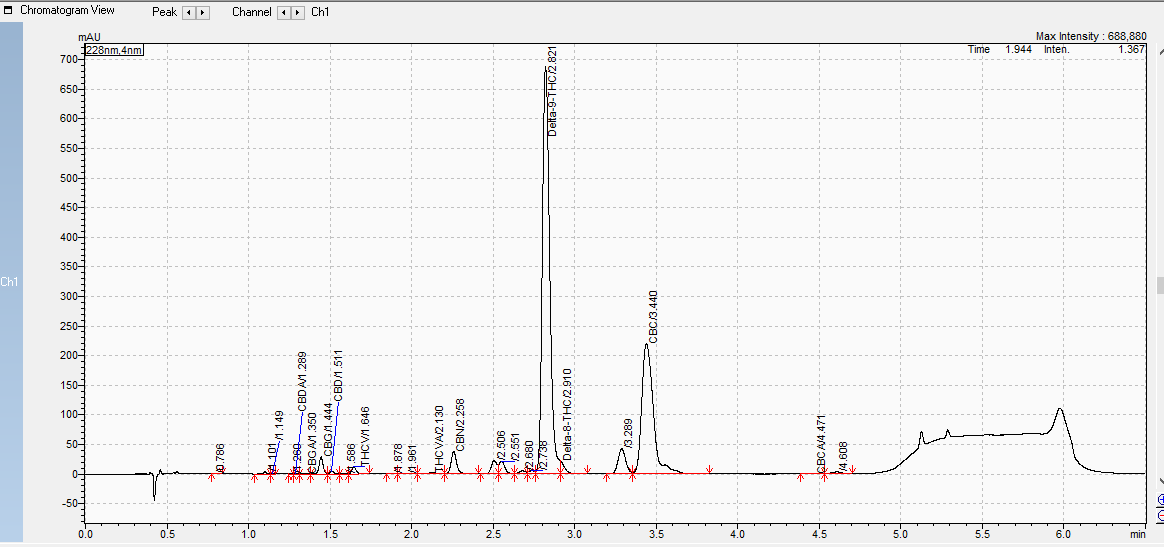
Been chasing ghosts and expending resources on this problem for a while now, any help again would be greatly appreciated! See chromatogram below for results of a batch with said undesired isomerization.
I’m of the opinion that it’s likely that it’s not actually isomerization, rather it’s coextracted compounds with nearly the same MW as the cannabinoids.
I tried pH adjustments, media filtration, LLE, as well as several distillation passes. Moderate success here and there, highest I ever got co2 distillate was 91.x%.
Same distillation parameters with BHO yielded 99+% oil.
Although in your case, that’s quite a bit of CBC, if that’s even what it is.
Retention time in LC is not molecular weight, you’re thinking of MS. Retention time is just relative to interactions between mobile and stationary phases.
Agreed that’s a hell of a CBC peak. Are they sure that peak isn’t Thca that might be remaining after decarb? At least the little tail to the right of the cbc peak is thca, but if their MP does not include acid (formic acid usually) the cbc and thca peaks can coelute and add up… Either way I’d expect to find some thca (seeing as there is cbda) in there but I’m not seeing it labeled.
As to isomerization, possibly some is happening but I’d expect to see more d8 if anything significant is going on, that little tail on d9 could just be from an overloaded column (ie they need to dilute it more). Although I could see those peaks to the left of d9 as some isomerization byproducts, still not that significant imo.
What is the on column ppm of that d9 peak for reference? If you know.
Was hoping you would pop on here, you seem to have a ton of experience with CO2 in general and these mystery peaks.
I think your opinion on coextracted compounds is likely accurate, it does better explain the inconsistency of results.
Any techniques you listed that you noticed yielded the most repeatable results? 85%-90% is our goal, we were consistently there until recently. No major changes.
Not sure on the on column ppm of the D9 peak. CBC is incorrectly labeled. Lab labeled it that for R + D purposes. Best they know about it is that it is a neutral cannabinoid.
Tight fractions on a wiped film granted the best results. Ph adjustments were negligible, gave an extra 1-2%. A couple medias I tried netted 4% once but I had to use heptane to achieve it.
carbonic acid, (H2CO3 ), a compound of the elements hydrogen, carbon, and oxygen. It is formed in small amounts when its anhydride, carbon dioxide (CO2), dissolves in water. … Rainwater infiltrating through the soil absorbs carbon dioxide from the carbon dioxide-rich soil and forms a dilute solution of carbonic acid
I’d dry the input material as much as possible. Carbonic acid is formed when co2 and water interact with each other.
pH is off. You’re creating these cannabinoids in distillation most likely. CO2 tends to do this. A lot of people refuse to use CO2 for this exact reason. Also if you scrub with T5 under hot conditions or have any left over in the flask this will happen. Could be a combination of both. I avoid CO2 at all costs… It’s the worst solvent possible for this process. There’s plenty of folks coming to this realization finally. (a second late and a penny short).
you arent seeing these cannabinoids in your biomass? assume they would concentrate significantly by the time they end up in your final oil.
what are the actual percentages of cannabinoids?
to determine if they are in fact converting you would need to mass balance your starting cannabinoid content to your final. potency of flower material, potency of spent biomass after extraction, finished oil, any waste along the way, etc etc.
my first thought once you eliminate these trace cannabinoids being in your starting flower, is what you already mentioned, your AC. i would recommend not using anything that is acid activated. steam activated AC will not cause isomerization. AC is a bitch to filter. 1 micron is not enough.
This looks like d10. Happy to help you or others with this problem try to avoid this. Sometimes it’s caused by a catalyst but not always. Send a DM here
If you have material that is isomerized I can also offer solutions to help possibly recoup your money.