Hi all,
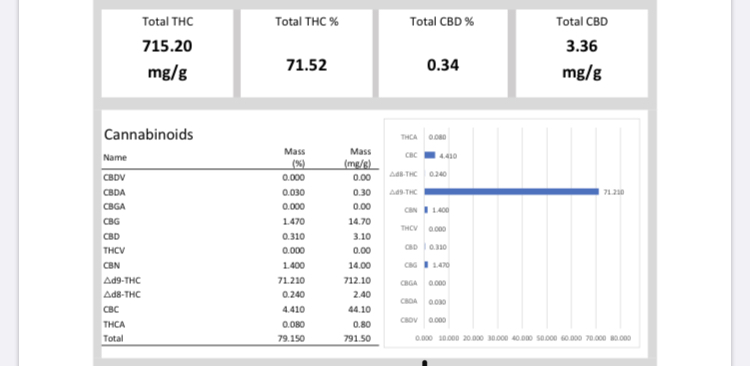
Looking for some help on my SPD process. Got results back for a first and second pass from the lab, and the results were not as great as I would have hoped😬. 1st pass was ~61% d-9 and ~66% TAC.
2nd pass from different starting material was ~71%d-9 and ~79%TAC.
Next round of testing, I will also be sending the crude, and everything from the same starting material. Crude, 1st and 2nd.
5L full bore system I pieced together, 45/50 head to 29/42 with a single e2m30 at the end.
Following the GLG first pass SOP, collecting mains from 165-185C while the system pressure at the pump sits between 30-50 micron.
Here is what we’re up to:
ISO extracted machine trim (I know I should be using cold etho, working on it).
Decarbed on the mag stirrer then scrubbed with T41 and degummed with DGE as per CarbonChemistry website SOP.
Dissolved in hexane and vacuum filtered. Then brine wash (10% salt by volume, distilled water) in sep funnels with low, high and neutral PH.
Evap hexane and redissolve in ISO to 10:1 and into the freezer for ~36hrs to -25C. (Can I sublimate dry ice to lower temps further? I don’t want it to throw off the PH or anything weird. Or chill my containers in a cooler/chest with the dry ice around it? I think it should be colder, especially with ISO instead of etoh.) Vacuum filter, evap, into the BF.
Then GLG SPD first pass SOP.
I have read here on the forum that there is a 2nd pass SOP on the GLG site but I don’t see it on the shared drive. Just two of the same versions of the first pass SOP and the Wiper SOP.
System pressure sat around 20micron or less at the pump during second pass I think but wasnt really sure where I should be collecting the main body during that second pass. The pressure being slightly lower, how should I tighten my collection tolerances in terms of the vapour temperature for the next run? The second pass I did, I just collected at the same temperatures (165-185) ![]()
but the WFE SOP on there makes me think i should try for 170-180 maybe?
What else can i be doing to help maximize potency without sacrificing yields unnecessarily?
Working on taking vapour temp during each run and graphing for reference. is it possible to pull data from LS bullseye vacuum gauge? there is a USB port, Non Bluetooth version.
Things on my list to get:
Ethanol
Ultra low freezer
Pressure filtration
Fraction finder
Here is the second pass. Doesn’t have a smell
or taste, I dunno.

Here is the first pass

TIA everyone