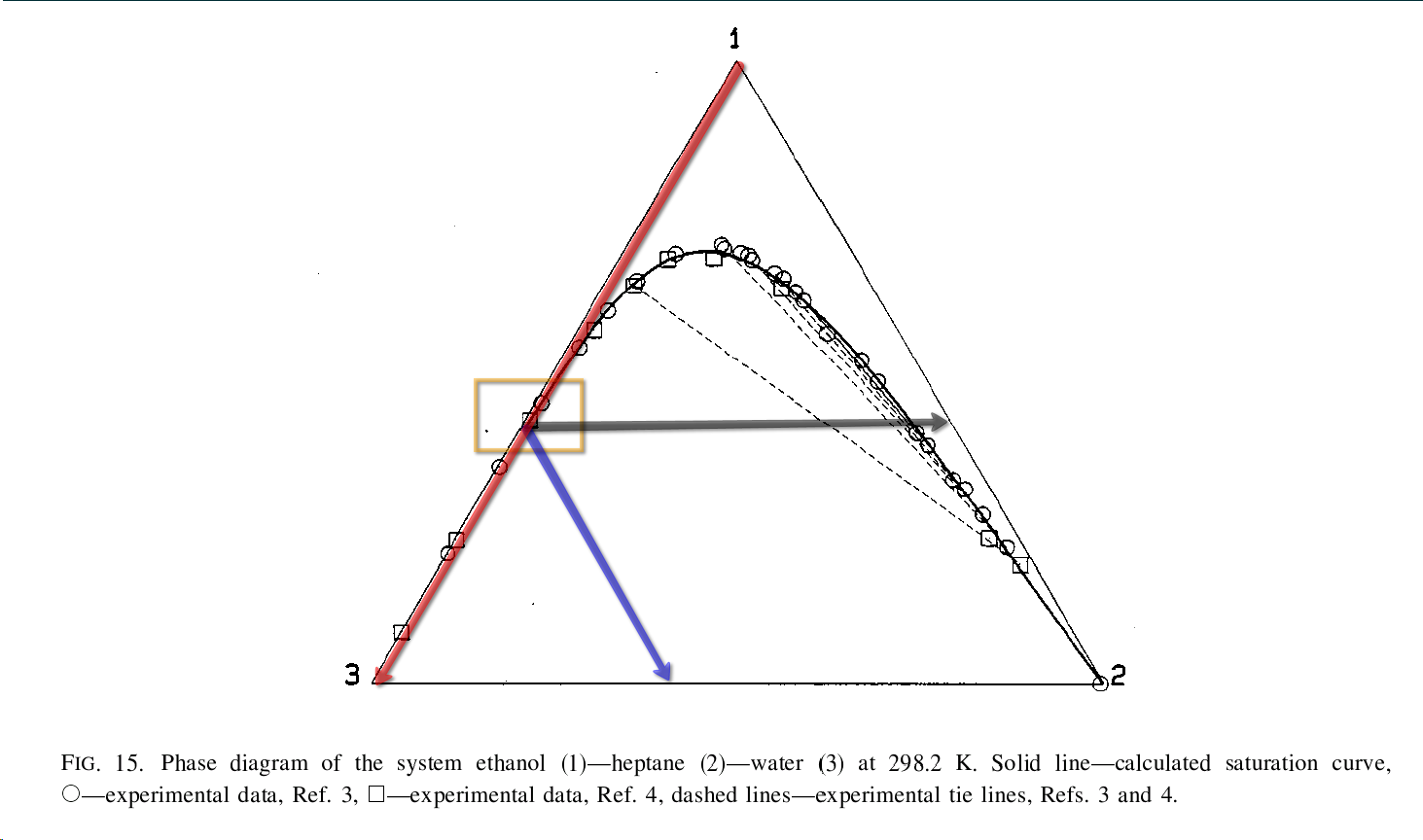
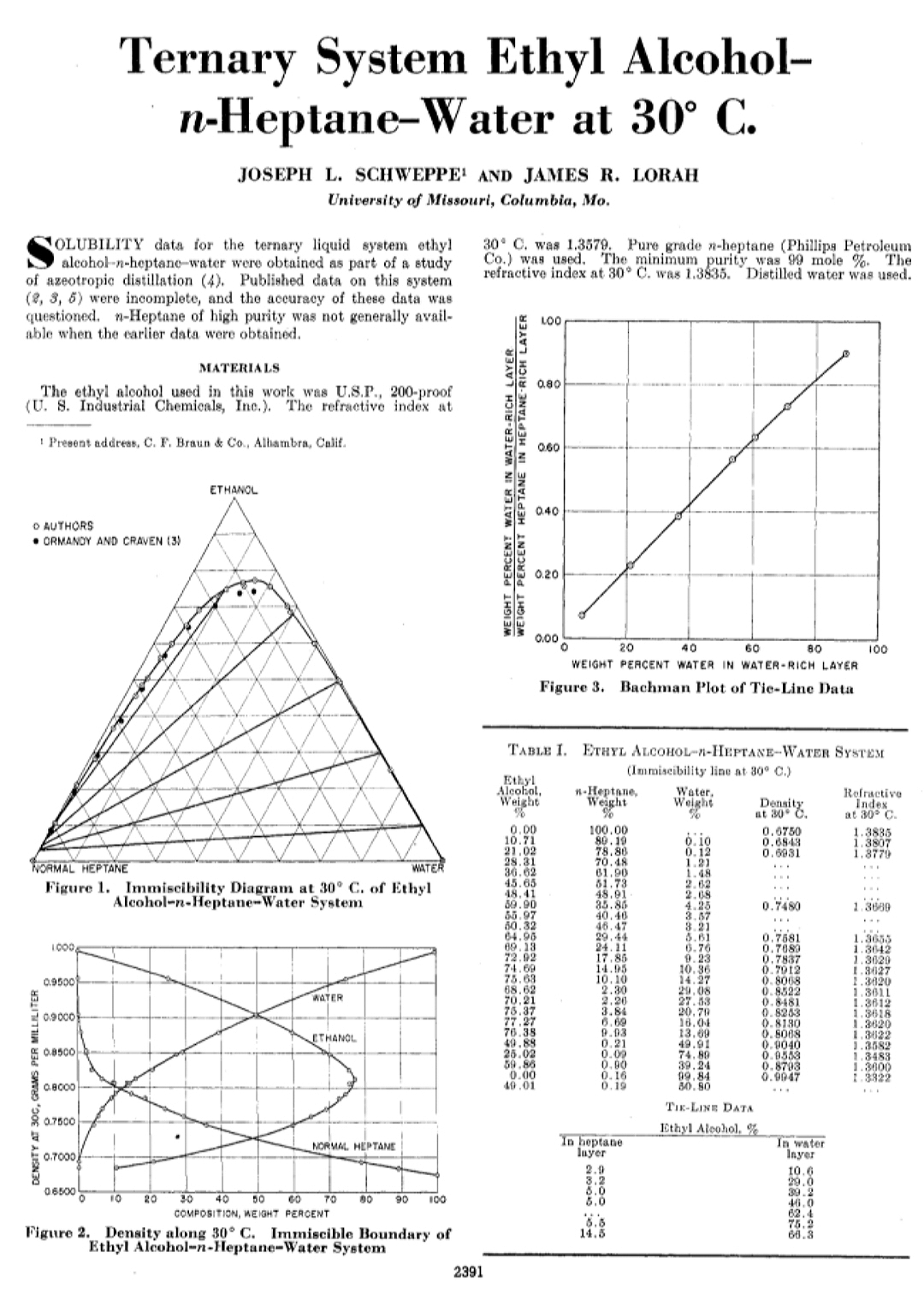
This excerpt adds a little more numerical detail to what you posted above,
@back2thefuture!

It should also clarify the actual behavior of the mixtures,
@sativum.
You are definitely on the right track, @back2thefuture, and thank you for the references! I am pleased to see you thinking critically about this problem, and in no way find your comments to be some sort of personal attack. I just assume positive intent, and I hope others do the same… especially because I or anyone else can accidentally sound like an asshole in plain text. I could very well be wrong in my hypothesis, and I certainly want to know if that is the case! It is only by honest reporting and thoughtful communication that we can find the truth, so more power to ya! 
You are certainly correct about the enormous disparity in percentages between the [heptane-denatured ethanol] sold for cannabis extractions and the [full binary (w/o water) or ternary (with water) azeotropes] described in the literature. It is also accurate that the boiling points of heptane:ethanol and heptane:ethanol:water azeotropes are lower than those of heptane, ethanol or water… i.e. these are “positive azeotropes.” So yes, it does stand to reason that an azeotropic fraction could be fractionally distilled from the bulk of the solvent… at least to obtain lower levels of heptane, since the [full azeotropes] require so much more heptane than 5%. There are at least 4 major things I can see to consider in doing this:
-
The general rule of thumb for efficient fractional distillation of 2 materials is that they should have at least 20°C difference in boiling points. This is because most compounds do not have a steep vapor pressure curve just below their boiling point. Iow, even when something is still 10°C below its boiling point, it is probably already evaporating pretty rapidly. So unless one is using a rather tall and tortuous rectification column, it can be pretty difficult to adequately separate 2 substances closer than 20°C in their boiling points.
-
Going by the same logic, it stands to reason that the 2 substances having close boiling points will evaporate in a ratio closer to 1:1, depending on their vapor pressures at the given temperature/pressure of the system. This means that, assuming you know and use the precise boiling point of the azeotrope and no higher, for every fraction of azeotrope you evaporate at its boiling point this way, a slightly smaller fraction of the higher boiling point material will also vaporize. This is why the rectifier needs to be so tall.
-
The amount of heptane in the heptane-denatured ethanol is not always accurately 5%! From what little I have seen so far, it would appear the heptane concentrations in this stuff vary between about 3% and 15%! Although it would be nice to have reagent grade accuracy in concentrations, it cannot legitimately be expected when paying industrial grade prices. To put it another way, I don’t know of any conventions or standards dictating how accurate an ethanol “denaturant” concentration must be.
-
Something that could work in one’s favor is the fact that this particular ternary azeotrope, when recondensed, separates into those 2 layers with very different compositions! If one keeps the distillation slow and fractionated enough to effectively separate azeotrope from the bulk liquid ethanol, or at least to obtain some sort of biphasic system in the resulting distillate, one will have most of the heptane in the upper layer of said distillate. Even if the distillate is one continuous phase at whatever temperature it exits the distillation, it is possible to chill that liquid to get it to separate into layers! I believe you touched upon this point, in fact. Unfortunately, this behavior does not bode well for people doing cold liquid solvent extractions…  @YakdOff
@YakdOff
The cold trap condensate is a very good indicator of how the liquid behaves during an extraction, @fresh.botanicals. You are seeing (smelling) the top layer, which has an extremely high concentration of pure n-heptane! See op.
You are also right to say that azeotropes are technically only a vapor phase phenomenon, but the practical upshot is that the liquid phase has those same relatively strong interactions between the molecules that cause the azeotropic behavior when it evaporates. My go-to example of this is the hydrogen bonding between ethanol and up to 5% water; one can fill a glass to the very brim with pure 200 proof ethanol, then add up to 5% water dropwise and it will never spill over… in fact, the volume in the glass will decrease!
The high concentration of ethanol, along with that last bit is one of 2 reasons I believe these [5% n-heptane-denatured ethanol] solutions are still very hygroscopic, considering that is such a strong characteristic of pure ethanol. The other reason is that I tested the relative densities of a fresh drum of n-heptane-denatured ethanol and an older drum of the same brand that had been opened and equilibrated with normal room air for over a week, both tested at room temperature within the same half hour. Note: Both samples were drawn from the bottom of the barrels, up through a hand operated piston pump…
The fresh stuff gave a hygrometer reading of “201 proof” indicating a fairly low density, as expected of pure ethanol with a bit of heptane in it.
The equilibrated stuff gave a hygrometer reading of exactly “190 proof”, indicating a higher density from the natural accumulation of water in the solution.
 Phase Diagram")