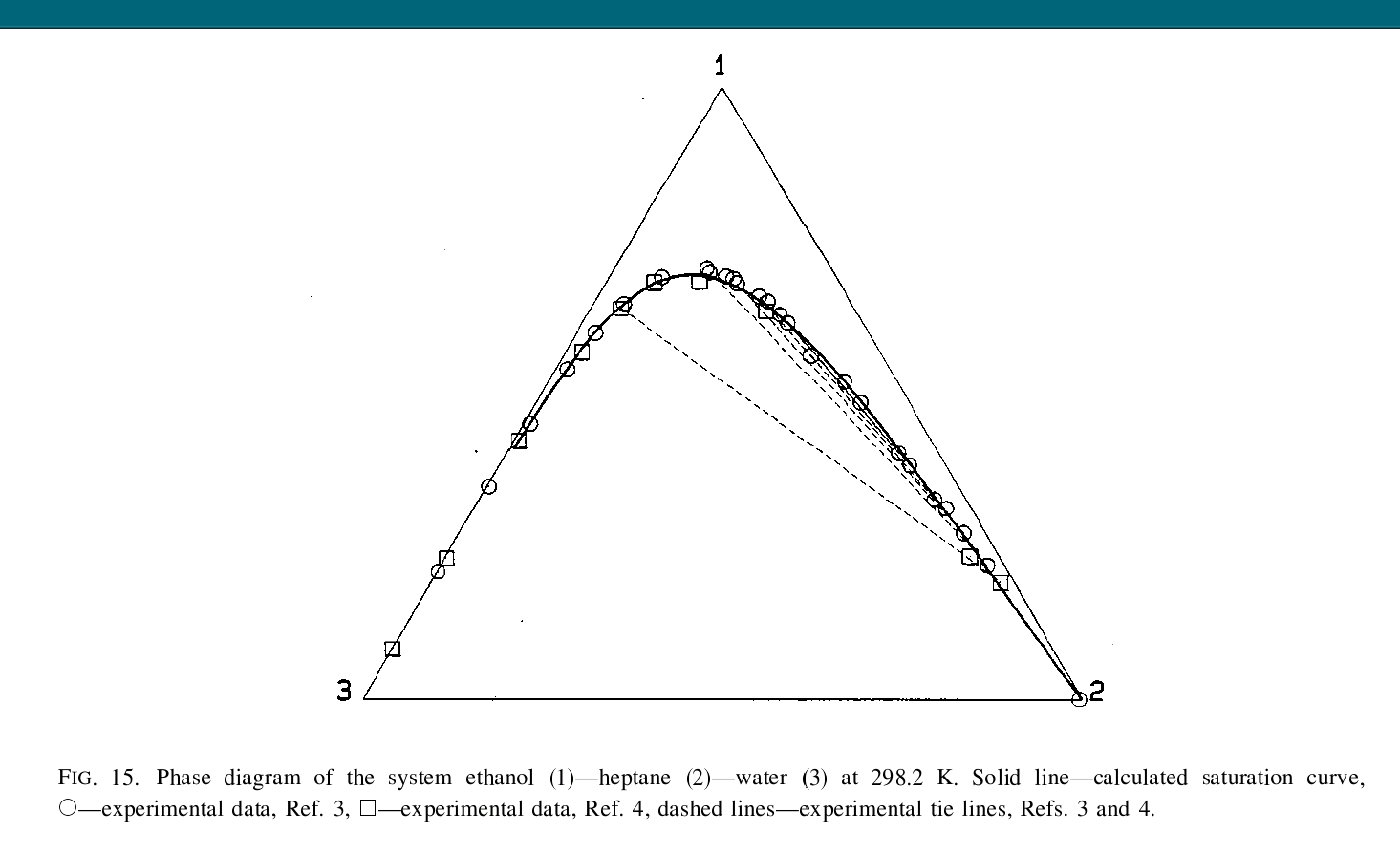
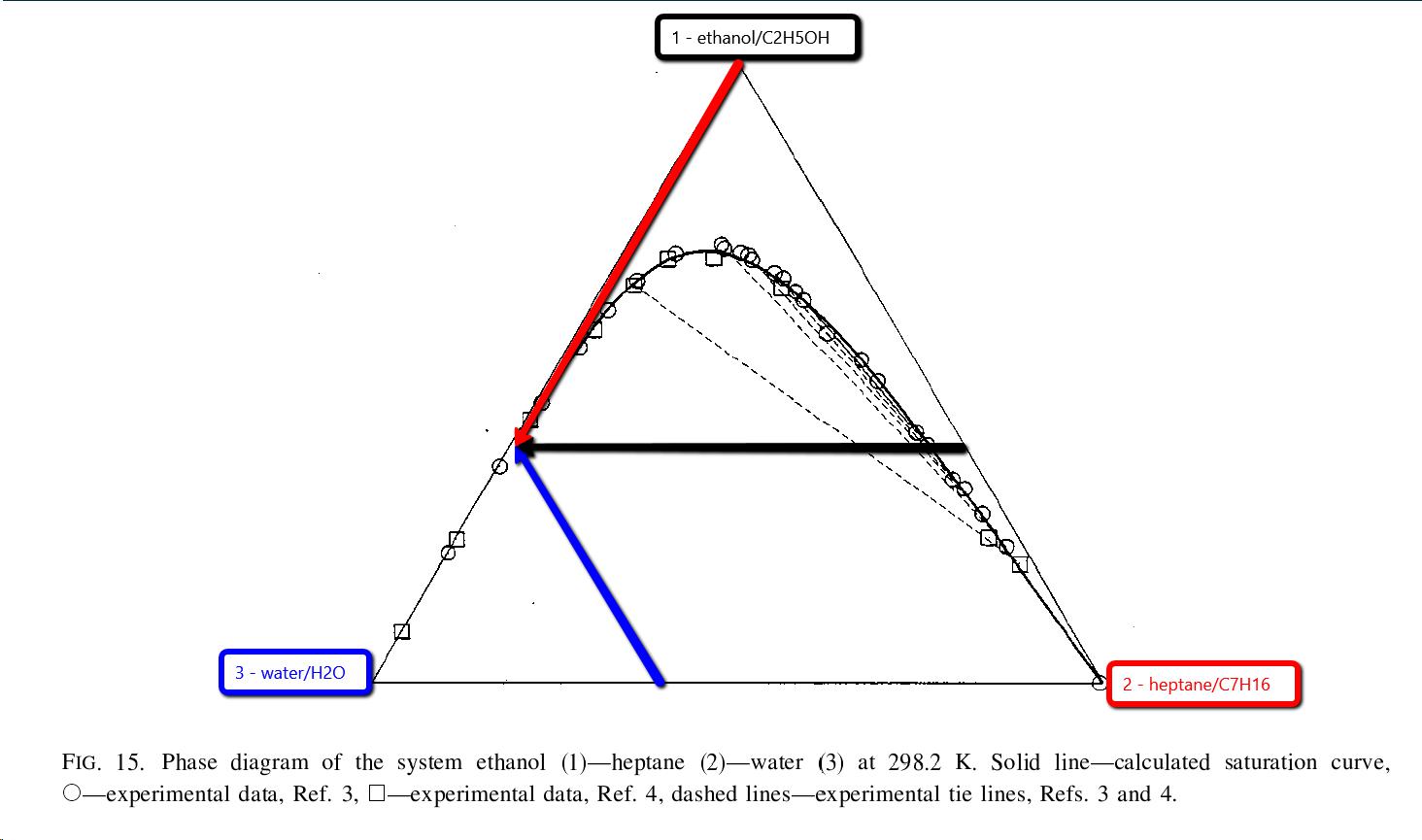
I’m wondering, what’s wrong with removing most of the heptane from denatured alcohol? Seems like nobody has given much attention to that question, unless there’s a different thread about it that I haven’t seen?
Fractionating columns are not exactly high tech or expensive to build from tri-clamp fittings. Nor would you need a large column that can distill many gallons per hour, either the binary or ternary azeotrope has a lot of heptane after all. With a decent reflux ratio and stainless scrubbers as packing in a 4-5’ long by 3" dia column, collecting maybe 1/2 to 3/4 gal/hr, I see no reason that wouldn’t pull out near azeotropic 60% heptane leaving very little behind. I’d add just slightly more water than is needed to satisfy the ternary azeotrope, but it would work without water.
Alternatively, maybe force it to separate into two phases and decant the upper phase? The problem here is then you’ll have a bunch of water ethanol mixture that needs to be reconcentrated, so it wouldn’t avoid the need for a column, and the whole volume of ethanol would need to run thru it rather than just the heptane rich fraction, so I see no advantage to this method.
But…
It may be possible to use something other than lots of water to cause phase separation, allowing one to use a normal recovery still to reclaim the ethanol after decanting the heptane. The only nonpolar extractant possibility I know of is silicone oil. Even heavy mineral oil is a bit too soluble to alcohol. From a manufacturer:
“Silicone fluid is highly soluble in hydrocarbon solvents such as toluene, xylene, ligroin, and mineral spirits as well as in chlorinated hydrocarbons. However, it is insoluble in ethanol , methanol, and water.”
It could be easily reused too. No doubt the cleaned up ethanol would need to be run thru a normal pot still, or rotovap to eliminate however much oil that did dissolve into it.
More commonly mentioned in literature is polar solvents (they’re working with much higher % heptane mixtures) and I found papers that show how a nontoxic deep eutectic solvent choline chloride+propylene glycol/glycerin and potentially a little water can work wonderfully for separating ethanol and heptane. Bulk livestock feed grade choline chloride is quite inexpensive too. It won’t let me post links yet, but if you’re interested you can piece these together. researchgate . net/publication/255771075_Deep_eutectic_solvents_as_extraction_media_for_azeotropic_mixtures
researchgate . net/publication/325221289_Effect_of_water_addition_on_extraction_ability_of_eutectic_solvent_choline_chloride_12-propanediol_for_separation_of_hexaneheptaneethanol_systems/amp
The liquid/liquid extraction style technique would also be even more useful to break the ternary azeotrope that’s collected from a fractionating still, in order to recover the heptane and little bit of ethanol as usable solvents. Much smaller volumes of deep eutectic solvent needed that way. Fractionation and then separate the azeotrope is probably how I’d go about it.
If one could find grades of denatured ethanol with methanol or some other less problematic solvents thatd be great but they don’t seem nearly as common or readily available. Nonetheless, buying heptane denatured stuff and fractionating off the heptane looks potentially much easier than fermenting and producing many gallons of azeotropic ethanol. I do have some experience with that… Its not a bad option either. If you do it, my recommendation is find some bulk wine in IBC totes for as cheap as possible and build stills (as many as you need) with beer kegs and 3" tri-clamp spools, packed with scrubbers. Don’t expect over 1 gal/hr of near 95% etoh from a still that size. They don’t have to cost too much, but you better insulate them and have an efficient fuel burning heat source (with a heat tranfer fluid hopefully) or cheap electricity. Its helpful to initially run the wine thru a pot still cranked up as fast as it will go, to “strip” the ethanol out as 40-60% before feeding it into the fractionating still. I’m assuming many of you are reasonably near wine country… 
If you’re lucky enough to find super cheap or free wine like I did, it won’t cost much more to produce the 'shine than denatured alcohol costs. Unless you value your time much. Having the equipment to produce drinkable spirits has its own set of risks and benefits though. I know of a guy down the road who got all the way thru the process of pressing/fermenting apples, building the still, producing the alcohol and then said “screw it, this stuff tastes too good to ruin with a bunch terpenes from trim”
Be aware it can be difficult to avoid some flavor from home made ethanol getting into the extract, whether that’s a problem depends on many factors.
I haven’t used it, but methanol is also intriguing in my opinion. Particularly if you’re already set up to perform extractions below -60 or your post processing steps can handle some extra hydrophilic contaminates.
P.S. I’ve been lurking and sometimes posting in forums on a wide range of subjects since around 2006, but this forum really stands head and shoulders above the rest! I’m really loving it and learning lots, so BIG kudos/congrats/all the rest to everyone who keeps this thing running and the folks contributing their experience and providing real usable high quality information.
Edit:
In regards to measuring the water content of heptane denatured ethanol. Seems like it would be feasible to use a hydrometer (although it would need to be able to read a bit lower SG than a normal 0-200 proof hydrometer) to get the SG of the fresh denatured 200 proof. Then take a sample, add a measured amount of water, record the new specifc gravity, maybe repeat that again just to be sure. At least until the phases separate.
I’d try it myself but I don’t have the right hydrometer. Maybe measuring the SG at freezer temperature could cancel out the lower density of the heptane to allow the normal hydrometer to work. Hmmm…
Edit 2:
Cool, that works. 95% EtOH 5% n-heptane measures exactly 190 proof (aka 0.8156-0.8160 SG) on my hydrometer, at -32°C. It was syphoned out of a freshly opened barrel and put directly in the freezer two days ago so there shouldn’t be much water.
So that’s a start. I’ll try adding some water to small amount and remeasure once its cooled off.