the above response suggests it’s NOT something you’ve actually done before. I don’t have a problem with that part…so far nobody proposing recarboxylation here has.
however, your initial statement implies more intimate knowledge of the synthesis than your second.
with 30 years of synthetic chemistry behind you, it is not unreasonable that you could be fairly sure a synthetic route was available without having actually performed the synthesis.
However, when someone explicitly asks if you’ve done this before, it’s not a brilliant idea in a forum like this to ignore that question (it speaks to credibility).
You then give a vague, hand waving description, of how it might be achieved. The functional groups you refer to have names. use them. the required synthetic reactions have names. use them. the tools required have names. etc…not doing so brings the claim of 30 years playing this game into question. you see that right?
IF you were selling/teaching this technique, keeping exact details to yourself would certainly be appropriate. Which is why I asked it that was the case.
Because you didn’t respond, the reader is required to guess. My guess is that you haven’t done it. Don’t sell it. And couldn’t perform the trick today without additional reading or R&D. Others are making even less charitable guesses. based on what you have, and have not said.
currently your “me too” post ads little to the discussion…and imo diminishes your credibility. which seems counter productive for a vendor or consultant.
in no way am I suggesting you refrain from commenting. nor am I claiming that your proposed synthetic route is bunk. You didn’t come close to providing enough information for me to draw that conclusion.
It’s beyond my skill level, and seems like success would only be good for bragging rights, but I’d put my money on it being possible to at least some extent.
As a geneticist, I’d also bet money that I could achieve an enzymatic re-carb if I mutagenised and selected.
Theres a chemist lurking around here that has successfully recarbed both CBDa and THCa but he’s being slippery about sharing the Tek… It is 100% possible, but also way above my head
Has anyone tried formylating THC using HMTA in TFA? I have used this on naphthols and phenols to install aldehydes. A subsequent Pinnick oxidation will yield the carboxylic acid. I know you all are trying to run this in large scales but at a smaller scale this would theoretically work. It doesn’t hurt to try. I am also aware of olefin isomerization under acidic conditions, but proof of concept is more important here.
Then, in 2011 Nadine Roth at the University of Freiburg in Germany published this paper where she also showed carboxylation of THC-A.
There are two isomers of THC known as THC-A and THC-B. Reactions regarding addition of a carboxylic acid group can be confusing due to the nomenclature here. Also, don’t confuse this with THC-Acetate either
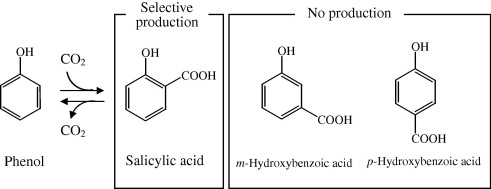
I’m curious to see how regioselective a Kolbe–Schmitt reaction would be when tried with THC.
This paper shows the ortho position being prefered by the -COOH.
Might be a good time to revive this thread. I have a few methods to try this on.
Using the phenoxide as a directing group to produce an aldehyde ortho to it and then oxidizing it to the acid.
Using the phenoxide with a reactive electrophile that will form a carboxylic acid upon workup.
Using a biphasic system in which CO2 is generated in situ and selectively added to the ortho position.
Note: I have used all three of these reactions on a naphthol scaffold and have only seen the electrophilic aromatic substitution form the ortho product.